Article Text

Abstract
Aims Following the development for liquid biopsies of the SiRe next-generation sequencing (NGS) panel that covers 568 clinical relevant mutations in EGFR, KRAS, NRAS, BRAF, cKIT and PDGFRa genes, in this current study, we apply this small NGS panel on tissue samples of lung cancer.
Methods A total of 322 specimens were prospectively tested. Technical parameters were analysed on both cytological and histological samples. In a subset of 75 samples, the EGFR SiRe results were compared with those generated by the European Community (CE)–IVD EGFR assay on Idylla platform. Clinical outcomes of 11 patients treated, on the basis of SiRe results, were also evaluated.
Results Only 28 (8.7%) specimens failed to produce a library; out of the 294 remaining samples, a total of 168 somatic mutations were found. In nearly all instances (74/75–99%), the EGFR SiRe results were confirmed by Idylla. In general, SiRe analytical parameters were excellent. However, histological and cytological specimens differed in relation to average reads for sample, mean number of mapped reads, median read length and average reads for amplicon. Treatment outcome evaluation in 11 patients showed a partial response in 82 % (9/11) patients with a median progression-free survival of 340 days.
Conclusions The small gene panel SiRe is a clinically relevant tool useful to widespread the adoption of NGS in predictive molecular pathology laboratories.
- NSCLC
- NGS
- predictive molecular pathology
Statistics from Altmetric.com
Introduction
During the last 10 years, a revolution in DNA sequencing strategies has taken place, with the development of massive parallel sequencing, also known as next-generation sequencing (NGS). This technology has first been applied to the research setting for the de novo genome sequencing by using broad gene panels, leading to development of advanced analytical and bioinformatic protocols, leading to a better understanding of the major pro and cons of this fascinating procedure.1–9
The introduction in clinical practice of affordable benchtop NGS sequencers decreased sequencing costs, moving NGS from a few large sequencing core centers to a much larger number of individual laboratories.10–13 Until recently, the Ion Torrent NGS platforms has mostly been employed to run extended panels containing from 50 to 409 cancer driver gene targets (eg, Ion AmpliSeq Hot Spot Cancer panel and Ion AmpliSeq Comprensive Cancer Panel).14 15 More recently, Lifetechnologies (Carlsbad) has released a CE–in vitro diagnostic (IVD) 22-gene target panel (Oncomine Ion AmpliSeq Colon and Lung Cancer Panel V.2) to encourage implementation of the NGS in predictive molecular pathology setting of patients with metastatic colorectal cancer (mCRC) and non-small cell lung cancer (NSCLC).16 17 As a general rule, small gene panels require a lesser abundant DNA input, which makes NGS analysis feasible even on small tissue samples.16 In particular, in most cases, NSCLC is diagnosed in the advanced disease stage by minimal invasive procedure, limiting the quantity of available DNA for molecular characterisation, which makes the use of large NGS gene panels problematic.18–20 Keeping this in mind, we have recently developed a narrow NGS panel (SiRe) covering 568 clinical relevant mutations in six genes (EGFR, KRAS, NRAS, BRAF, cKIT and PDGFRa) involved in NSCLC, gastrointestinal stromal tumour, colorectal cancer (CRC) and melanoma.21 In previous studies of ours, the SiRe panel analytical performance was assessed on cell line DNA and by using an artificial reference standard with multiple mutations, and then applied to the analysis of liquid biopsies and circulating tumour DNA (ctDNA), showing that SiRe is a feasible alternative to real-time PCR-based assay to mutational status assessment on ctDNA.21 22 To date, the NGS SiRe panel has not been applied to histological and cytological specimens; thus, in this current study, we expanded our research to apply the SiRe panel, with a dedicated variant caller parameters file, on tissue samples, investigating the advantage of a small panel in a routine practice of a molecular pathology laboratory to select patients with NSCLC for target treatments.
Materials and methods
Patients and samples
From January to December 2016, a total of 322 NSCLC samples were prospectively tested by using SiRe NGS panel; a single sample of a given tumour site was tested for each patient. Patients and samples characteristics are summarised in table 1A–C. In particular, 142 (44.1%) were histological and 180 (55.9%) were cytological specimens. In detail, most of the histological samples were small biopsies (n=107, 75.4%), while only in 35 (24.6%) cases a surgical resection was available. As far as cytological preparation are concerned, 135 cases (75.0%) were direct smears, and 45 (25.0%) were cell blocks. As far as cytological specimens type are concerned, 157 (87.2%) were fine needle aspirates (FNA), 6 (3.3%) were brushing and 17 (9.5%) were effusions. Regarding the tumour site, 87 (27.0%) samples were collected from metastases (44 (50.6%) were histological and 43 (49.4%) were cytological). A total of 235 (73.0%) samples were collected from primary tumour (98 (41.7%) were histological and 137 (58.3%) were cytological). Considering the pathological reports, standardised according to the eighth edition of WHO classification, morphological diagnosis were as follows: adenocarcinoma (ADC; n=181; 56.2%) diagnosed only on morphological grounds; NSCLC not otherwise specified (NOS) refined by immunohistochemistry into favour ADC (n=101; 31.4%); squamous cell carcinoma (n=6; 1.9%), neuroendocrine carcinoma (n=4; 1.2%) and NOS (n=30; 9.3%).
Variant caller parameters file for variant calling plug in 5.0 tissue adapted
Variant caller parameters file for variant calling plug in 5.0 tissue adapted
Variant caller parameters file for variant calling plug in 5.0 tissue adapted
SiRe panel and DNA purification
As reported in our previous study, a single primer pool customised panel leading to the selection of 42 amplicons (ranging from 125 to 175 bp) targeting six genes (EGFR, KRAS, NRAS, BRAF, cKIT and PDGFRa) associated with treatment outcome in N S CLC, GIST, CRC and metastatic melanoma (whose acronym is SiRe), was designed by using the Ion AmpliSeq Designer suite V.5.3.1 with hg19 as reference genome.21 22 The amplicons design covering 5.2 kb of genomic DNA was optimised for the simultaneous analysis of 16 samples with the 316v2 chip (Thermofisher, Foster City, California, USA) on a Personal Genome Machine (Thermofisher).21
In relation to the extraction modalities, for both histological and cytological samples, after a visual inspection by a fully qualified pathologist (EV), a neoplastic area with more than 5% of neoplastic cells was selected. Cells were scraped by using a sterile scalpel from the slides (after cover-sleep removal in xilene only for cytological samples), and DNA extraction was performed with QIAamp DNA Mini Kit (Qiagen, Crawley, West Sussex, UK), following the manufacturer instructions, resuspending the DNA in 30 µL of RNAsi/DNAsi free water (Ambion, Thermofisher, USA). To evaluate the quantity (ng/μL) and quality (in term of DNA Integrity Number (DIN)) of extracted DNA, 1 µL of resuspended DNA for each sample was analysed by using genomic DNA screen-tape assay on the 4200 TapeStation system (Agilent Technologies, Santa Clara, California, USA) with a proprietary software, prior to proceed to NGS library preparation.
SiRe panel NGS library preparation and sequencing analysis
Starting from 10 ng of genomic DNA, by using SiRe panel, libraries were prepared and purified on the Ion Chef automatic platform (Thermofisher), and eight samples were added per run. Libraries generation were carried out on Ion Code plates and amplified using Ion AmpliSeq DL8 Kit (Thermofisher). Then, under the thermal conditions defined by the manufacturer, we used 22 cycles for amplification and six cycles for library reamplification after barcoding. Purified and combined libraries from two Ion Chef runs, derived from 16 patients, were diluted to 70pM. The pooled libraries were reloaded into the Ion Chef instrument, and templates were prepared by using the Ion PGM Hi-Q IC Kit (Thermofisher). Finally, templates were loaded into the 316v2 chip and sequenced on PGM.
In any single case, signal processing and base calling were carried out using the default base-caller parameters on Torrent Suite (V.5.0.2), and coverage analysis was performed using SiRe-designed bed files with coverage plug-in (V.5.0.2.0). The bioinformatic pipeline based on the SiRe Variant caller plug-in (V.5.0.2.1) parameters, enabled for automatic variant calls; the threshold parameters were specific optimised for tissue based diagnostic (online supplementary table 1A). Only variants with >20X allele coverage and a quality score >20, within an amplicon that covered at least 500X alleles, were called. The detected variants, for any SiRe panel covered gene, were also considered in relation to sample type (histological vs cytological), collection modalities (biopsy vs surgical resection; FNA vs brushing/effusion) and the percentage of neoplastic cells, categorised in three classes (5%–25%, 26%–50% and >50).
Supplemental material
SiRe analytical performance evaluation
In order to evaluate analytical performance of SiRe panel on NSCLC routine samples, the following run metric parameters for each patient were analysed: number of total reads, number of mapped reads, mean reads length, percentage of reads on targets, mean coverage per amplicon and uniformity of coverage. Each one of these parameters was also considered in relation to sample type (histological vs cytological), collection modalities (biopsy vs surgical resection; FNA vs brushing/effusion) and, only for cytological samples, in relation to sample preparation (direct smears vs cell blocks).
Idylla assay
In addition,the Idylla fully automated platform (Biocartis, Mechelen, Belgium) was used to verify SiRe panel results.23 24
The samples inclusion criterion for this comparative analysis was the availability of at least 10 µL of archival DNA. In particular, 75 samples (44 with a SiRe EGFR mutated and 31 with a SiRe wild-type results) were selected. In any case, the Idylla EGFR genotyping workflow was carried out as previously described to assess the concordance rate between our NGS approach and the Idylla CE-IVD-validated assay.
Treatment response evaluation basing on SiRe results
In accordance with the oncologists of two different South Italy institutions (A.O.R.N. Vincenzo Monaldi and A.O.R.N. Antonio Cardarelli), to evaluate the clinical efficacy in term of treatment response prediction of SiRe panel, clinical outcome in patients undergoing to first or second generation of anti-EGFR tyrosine kinase inhibitors (TKIs) was evaluated. To this aim, we selected patients according to the following criteria: (1) evidence of exon 18–21 EGFR mutation, (2) administration of target treatment during the course of the disease and (3) the availability of at least 3 months of follow-up. Eleven patients met these inclusion criteria (five men and six women; median age, 66.3 years). In particular, five patients with NSCLC were treated with gefitinib and six patients with afatinib; among these patients, seven were selected starting from cytological specimens derived DNA and four starting from histological samples derived DNA.
Disease status was evaluated in all patients by total body CT scans. The response rate was evaluated based on Response Evaluation Criteria in Solid Tumors (RECIST V.1.1) guidelines. Progression-free survival (PFS) was calculated from the start of target treatment to the date of confirmed progression or death from any cause. PFS data were plotted as Kaplan-Meier curve. Written informed consent was obtained from all patients and documented in accordance with the general authorisation to process personal data for scientific research purposes from ‘The Italian Data Protection Authority’ (http://www.garanteprivacy.it/web/guest/home/docweb/-/docwebdisplay/export/2485392). All information regarding human material was managed using anonymous numerical codes, and all samples were handled in compliance with the Helsinki Declaration (http://www.wma.net/en/30publications/10policies/b3/).
Statistical analysis
Data are presented as number and percentage for categorical variables, and continuous data are expressed as mean±SD, unless otherwise specified. The χ2 test and Yates’s continuity correction or Fisher’s exact test were performed to compare the differences between two percentages or proportions. Particularly Fisher's exact test was used where the χ2 test was not appropriate. Instead, to evaluate significant differences of means, we performed a Student’s t-test. Analysis of variance (ANOVA) test was used in multicomparison among means. When ANOVA test was positive (p values <0.05), pairwise comparisons were performed with Scheffé’s test. Finally, analytical parameter correlation to sample characteristics were reported as Cartesian plans, while PFS data were plotted as Kaplan-Meier curve. We considered all statistical tests with p value <0.05 to be significant. All data were analysed with Matlab statistical toolbox V.2008 (MathWorks, Natick, Massachusetts, USA) for 32-bit Windows (online supplementary table 1B).
Supplemental material
Results
DNA quantity and quality evaluation
Prior to proceed to NGS library preparation, by using genomic DNA screen-tape assay on the 4200 TapeStation system with a proprietary software, the quantity (ng/μL) and quality (DIN) of each sample was evaluated, and results are reported in online supplementary table 1C. On the overall (n=294), the average quantity of DNA was 45.9 ng/µL, and the mean DIN was 5.2. No significant difference in DNA quantity and quality were obtained taking into account the sample type (histological (43.1 ng/µL; 5.3) vs cytological (48.0 ng/µL; 5.0)), collection modalities (biopsy (37.0 ng/µL; 5.3) vs surgical resection (61.7 ng/µL; 5.2) and sample preparations (direct smears (55.2 ng/µL; 5.1) vs cell blocks (26.5 ng/µL; 4.9)) for cytological specimens. The only significant difference (p values<0.0001) was observed between FNA (32.9 ng/µL; 5.0) versus brushing/effusion (143.3 ng/µL; 6.5).
Supplemental material
SiRe analytical performance
On the overall, 322 analysed samples, 28 (8.7%) failed to produce an adequate library. In relation to run metric parameters, SiRe showed an average of 166 206.91 reads per sample with a median read length of 130.64 bp. The mean number of mapped reads per sample was 164 941.58 with 96.23% reads on selected targets. Regarding the coverage, each amplicon showed an average reads of 3813.85 and a uniformity distribution on the 42 amplicons of 97.79%.
As reported in Materials and Methods section, each one of these run metric parameters was also considered in relation to sample type (histological vs cytological), collection modalities (biopsy vs surgical resection; FNA vs brushing/effusion) and, only for cytological samples, in relation to sample preparations (direct smears vs cell blocks). The results are reported in table 2A–C and graphically showed in figure 1. More in detail, between histological and cytological samples, there were a significant differences for Average reads for sample (149 217.18<179 674.38, p values=0.0379) for Mean nr. of mapped reads per sample (147 739.80<178 577.14, p values=0.0435), for Median read length bp (129.88<131.24, p values=0.0009) and for Average reads for amplicon (3382.14<4158.16, p values=0.0236). Between biopsy and surgical resection, there was a significant difference only for Median read length bp (130.48>128.18, p values=0.0190).
SiRe run metric parameters in relation to sample characteristics
Sample type in relation to SiRe run metric parameters
Cytological sample preparations in relation to SiRe run metric parameters

Run metric parameters graphically reported in relation to sample type (histological vs cytological) and collection modalities (biopsy vs surgical resection; FNA vs brushing/effusion). FNA, fine needle aspirate.
SiRe sequencing results
Only variants with >20X allele coverage and a quality score >20, within an amplicon that covered at least 500X alleles, were called. On the total analysed patients (n=294), a total of 168 somatic mutations were found; eight patients harboured two concomitant EGFR mutations; ttwo KRAS concomitant mutations were detected in three patients; one patient harboured a concomitant EGFR and KRAS mutation; and one patient showed a concomitant KRAS and BRAF mutation. The specific types of concomitant mutations detected in each patient are reported in table 1A–C. In particular, 58 (34.5%) were EGFR, 95 (56.6%) were KRAS, 1 (0.6%) was NRAS and 14 (8.3%) were BRAF. All the PDGFR (n=65) and c-KIT (n=48) detected alteration were polymorphic variants (as reported in cosmic database; www.cosmic.it, last access 12 July 2017) and were excluded from the analysis. In table 1A–C, the detected mutations are reported, for any SiRe panel covered gene, in relation to sample type (histological vs cytological), collection modalities (biopsy vs surgical resection; FNA vs brushing/effusion) and the percentage of neoplastic cells, categorised in three classes (5%–25%, 26%–50% and >50%). We observed a significant difference only between 5%–25% and >50% (28.57%<39.46%, p values=0.007).
Idylla assay results
As specified in Material and Methods section, 75 samples were also analysed by Idylla automatic platform. In particular, 44 EGFR mutated and 31 EGFR wild-type by SiRe panel were retrieved following the inclusion criterion. In 99% (74/75) of samples, the EGFR SiRe results were confirmed by using the CE-IVD EGFR mutational test on fully automated Idylla platform. Only one sample, with 5% of neoplastic cells, was not confirmed by Idylla platform for a 21 exon point mutation. Moreover, NGS identified two simultaneous clinical relevant mutations in four samples (n=2 ELREA/T790M, n=1 L861Q/G719A, n=1 L858R/T790M). Idylla system was able to find concomitant mutations in three out of four patients. In one patient, Idylla detected only the ELREA deletion missing the T790M point mutation (table 3).
Comparison between NGS data and Idylla results on NSCLC samples
Treatment response of patients selected by the SiRe panel
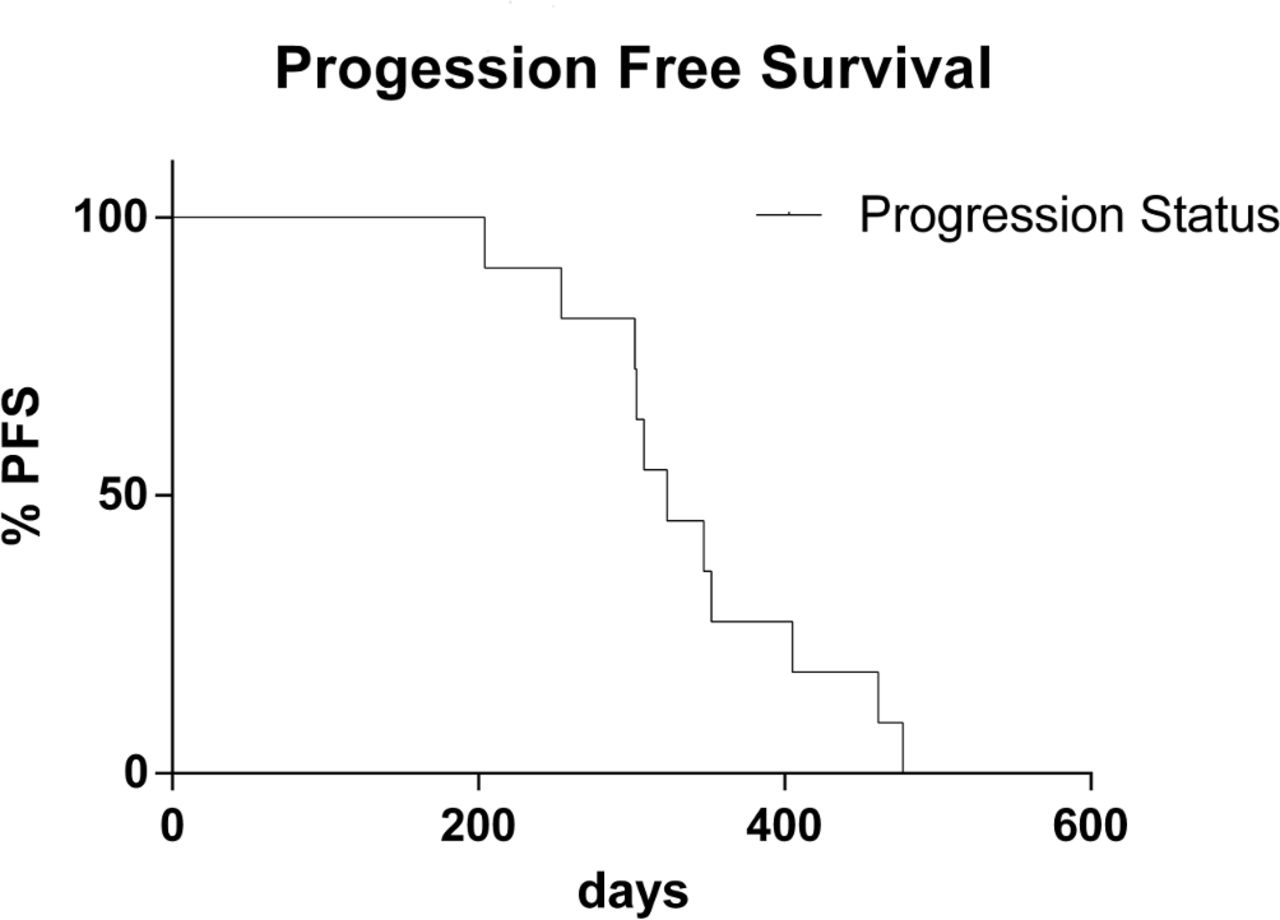
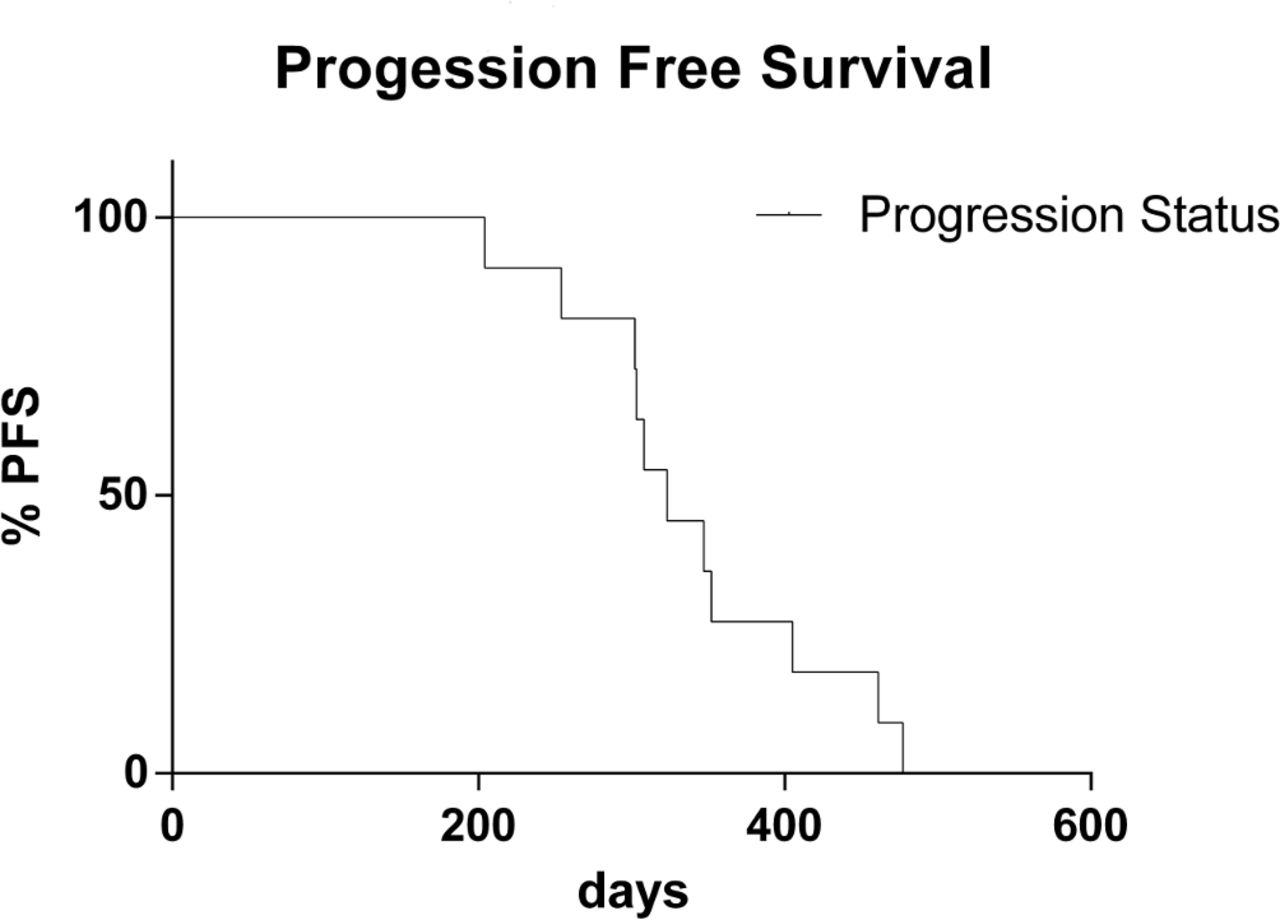
As specified in Materials and Methods section, 11 patients met inclusion criteria for treatment outcome evaluation (five men and six women; median age, 66.3 years). In particular, five patients with NSCLC were treated with gefitinib and six with afatinib. In 9 out of 11 patients, a partial response to treatment was observed (82%). No response was observed in two patients (18%). The disease control rate (objective responses plus stable disease) was 100% (table 4). In relation to survival, the median PFS was 340 days (range, 204–477 days) (figure 2).
EGFR TKIs treatments and clinical outcome parameters
{kind=link}
{kind=link}
PFS data plotted as Kaplan-Meier curve calculated from the start of target treatment to the date of confirmed progression or death from any cause. PFS, progression-free survival.
Discussion
In this study, we show that the performance of SiRe NGS panel on DNA extracted from NSCLC routine samples is excellent; thus, the SiRe panel can be used for mutational assessment of scant cellularity samples, such as cytology or small histological biopsies. SiRe is designed to be a small gene panel (5.2 kb) focused on biomarkers that are currently used in the clinical setting covering up to 568 clinical relevant mutations in six different genes (EGFR, KRAS, NRAS, BRAF, cKIT and PDGFRa).21 22 This approach, unlike wider genome analysis, is particularly useful in NSCLC molecular diagnostic setting; in fact, NSCLCs are diagnosed in advanced stage and in more than 80% minimal invasive procedure must be employed to obtain diagnostic materials. Therefore, limiting the quantity of available DNA for molecular characterisation by adopting a less broad NGS panel may be crucial in the predictive molecular pathology setting.25–27
Until recently, the Ion Torrent NGS platforms has employed broad commercial gene panels containing from 50 to 409 cancer driver gene targets (eg, Ion AmpliSeq Hot Spot Cancer panel and Ion AmpliSeq Comprensive Cancer Panel, respectively).14 15 More recently, Lifetechnologie has also released a CE-IVD 22-gene target panel (Oncomine Ion AmpliSeq Colon and Lung Cancer Research Panel v2) to encourage implementation of the NGS in predictive molecular pathology setting of patients with mCRC and NSCLC.16 17 As a matter of the fact, commercially available panels have some limitations, in particular when applied to paucicellular samples. In their experience, Leichsenring et al showed the feasibility to adopt cytological samples for NGS analysis with wide commercially available panels, as an important tool to evaluate the tumour evolution. This small cohort of patients (n=20) with matched cytological and histological specimens showed discordances in five cases; in particular, in three samples, the mutation was identified only in the histological specimens and two samples showed discordant mutation, respectively, on histological and cytological samples. These discordances should be related to the lower abundance of tumour cells (<10%) in effusion material than the other samples.28 In a larger cytological cohort (n=207) adopting the AmpliSeq Cancer Hotspot V.2 panel (50 genes), there was a 21% failure rate due to either a low DNA yield or a template/library preparation failure.29
Keeping this in mind, in this study, we have analysed the performance of SiRe panel and of its tissue-adapted variant caller parameters file on NSCLC routine samples. Out of a total of 322 cases, only 28 (8.7%) failed to produce an adequate library. As reported in results section, the analytical performance of SiRe Panel was more than satisfactory showing an average of 166 206.91 reads per sample with a median read length of 130.64 bp. In addition,the NGS workflow allowed a very cost-effective batching of samples (16 per run on 316v2 chip), regardless the type of tumour and the pathological preparations. As a result, turnaround time (TAT) can be as short as three working days, as recommended by international guidelines and, in addition, the recently developed Ion Chef automated library preparation station, which has a better procedure reproducibility and standardisation than manual procedures, also contributes to the short TAT. Therefore, the SiRe NGS panel can be a suitable solution in those clinical institutions that are performing molecular analysis only for standard of care actionable biomarkers, whereas larger cancer care comprehensive centres may be involved in performing larger NGS analysis to evaluate additional biomarkers for clinical trials patients’ enrolment. Thus, the SiRe seems to fill in an intermediate space between large panels and PCR-based assays, underlining the concept that NGS is a versatile technology, whose aim and scopes greatly differ among institutions and clinical setting. The reliability of the NGS SiRe panel is also demonstrated by the similar performance with that accomplished by the CE-IVD Idylla assay, underlining the robustness of this in-house validated approach. Moreover, as far as clinical treatment data are concerned, in this study, we demonstrated that the very majority of patients (82%), treated by EGFR TKIs basing on SiRe genotyping, showed a clinical benefit in line with data of principal clinical trials.30–32
In conclusion, our data showed that SiRe NGS panel, with tissue adapted variant caller parameters file, represents a robust and fast analytical tool for a large volume laboratory enabling the possibility to test tissue samples of NSCLC patients to assess EGFR and other clinical relevant genes mutational status for TKIs treatments administration. Currently, a multicentre prospective study is taking place to verify the performance of this simplified NGS workflow in different laboratories.
Take home messages
Small next-generation sequencing (NGS) gene panels require a lesser amount of DNA input; this makes NGS analysis available even on small tissue samples.
SiRe is a narrow NGS panel covering 568 clinical relevant mutations in six genes (EGFR, KRAS, NRAS, BRAF, cKIT and PDGFRa) involved in NSCLC, gastrointestinal stromal tumour, colorectal cancer and melanoma.
Here, we showed that SiRe panel represents a robust and fast analytical tool to test tissue samples of patients with non-small cell lung cancer in order to assess EGFR and other clinical relevant genes mutational status for targeted treatments administration.
Abstract translation
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.References
Footnotes
FP and CDL contributed equally.
Handling editor Professor Runjan Chetty.
Contributors FP, CDL, GT and UM conceived the study and wrote the paper. RiS, RoS, MN, MR and GG performed the experimental part. PP, EV and ClB contributed as pathologists. DR, CiB and FA contributed as oncologists. NS performed the statistical analysis. All Authors approved the final version of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained
Provenance and peer review Not commissioned; externally peer reviewed.