Article Text

Abstract
Paraneoplastic leukemoid reaction (PLR) is the extreme leukocytosis that occurs due to a non-haematolymphoid cytokine-secreting tumour (CST) in the absence of bone marrow infiltration by that solid tumour. The clinical presentation is widely variable, and therefore challenging. If the underlying malignancy is not clinically apparent, PLR could be mistaken for myeloproliferative neoplasms, altering the patient’s management. CSTs are highly aggressive tumours associated with a poor prognosis due to multiple mechanisms. Localising and treating the underlying malignancy is the mainstay of treatment. Both the treating clinician and the pathologist should keep a high level of suspicion for this entity in patients having unexplained leukocytosis. We herein discuss the underlying mechanisms, clinical presentation, pathological features, differential diagnosis and prognosis of this rare entity. An emphasis on the role of the pathologist is provided since the lack of knowledge on this entity can lead to dramatic effects on the patient, including unnecessary diagnostic testing and treatments.
- cancer
- leukocytes
- chronic myeloid leukaemia
- cytokines
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Paraneoplastic syndrome can present in a variety of haematological clinical disorders, such as anaemia, hypercalcaemia, erythrocytosis, granulocytosis and thrombocytosis.1 Lung cancer is thought to be the most common malignancy associated with paraneoplastic syndromes.2
Leukemoid reaction (LR) is defined as persistent leukocytosis (white blood cell (WBC) above 40 000/µL) in the absence of a haematologic malignancy.3 LR can be caused by infections, intoxications, malignancies, severe haemorrhage or acute haemolysis.3 Paraneoplastic leukemoid reaction (PLR) can be defined as the LR which occurs due to the presence of a non-haematolymphoid cytokine-secreting tumour (CST) in the absence of bone marrow infiltration by that solid tumour.4
The first PLR due to CST was reported in 1977 in a patient with lung cancer.5 Since then, it has been reported in patients with melanoma, mesothelioma, carcinomas and sarcomas of various origins (biliary tree, oesophagus, gallbladder, head and neck, liver, stomach, urinary bladder and thyroid).1 4 6–17
Underlying mechanisms of PLRs
PLR usually occurs in the setting of a CST. The most commonly secreted cytokine is granulocyte colony-stimulating factor (G-CSF). However, other cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-1a, b, IL-3, IL-6 and tumour necrosis factor (TNF)-α have been also reported.2 17 18 G-CSF is a naturally occurring glycoprotein that stimulates the proliferation and maturation of marrow progenitor cells into fully differentiated and functionally activated neutrophils.19 Normally, G-CSF is produced by vascular endothelial cells, fibroblasts, monocytes and macrophages. In patients with PLR, G-CSF is directly secreted by tumour cells into the host’s circulation, leading to cytokine-mediated granulocytosis.20 Some tumours can simultaneously secrete parathyroid hormone-related protein in addition to CSF, leading to combined hypercalcaemia and leukocytosis of the host.16
The tumour’s ability to secrete CSFs can develop simultaneously with tumour development (ie, with the primary clone/generation of tumour cells); or it can be secondarily acquired through subsequent dedifferentiation of the primary tumour.14 18 This ability can also be acquired in metastatic sites even if the primary tumour is not a CSF producer.18 Also, in tumours with multiple metastatic sites, CSF-secreting ability can be found in some metastatic sites while not found in the others. CSF-producing metastatic foci grow more rapidly than the non-CSF-producing metastatic foci of the same primary tumour.21 This can be explained by the phenomenon that CSF-secreting tumour cells can express CSF receptors on their cell membranes, which bind the same ligand that they secrete.11 This mechanism allows for autocrine growth induction of some CSTs. In addition to stimulating bone marrow myelopoiesis, CSTs can also induce a ‘qualitative’ effect by inhibiting myeloid cell differentiation in the tumour vicinity. This, in turn, leads to accumulation of immature myeloid cells which are called myeloid-derived suppressor cells (MDSCs). The term ‘suppressor’ is given as these cells have an immune suppressive effect which shields the tumour from being attacked by the host. It has also been found that MDSCs play an important role in inducing tumour angiogenesis.22
Clinical presentation
Clinical presentation of CSTs can be widely variable, and therefore challenging. Patients usually present with fever and/or symptoms related to the underlying malignancy.4 14 If the underlying malignancy is not clinically apparent, tumour-induced leukocytosis could easily be mistaken for infection or myeloproliferative neoplasm (MPN).23 This, in turn, can lead to unnecessary diagnostic procedures, such as bone marrow aspiration and biopsy with extensive molecular and ancillary testing.4 15 21 The presence of fever can distract the treating clinician’s attention to pursue a diagnosis of infection instead of PLR as the cause of unexplained leukocytosis. Yet, infection must be first excluded, not only because it is more common than PLR as a cause of secondary leukocytosis24 but also because the presence of active infection can be a contraindication for treating the underlying malignancy, particularly if chemotherapy is to be used.
The WBC count in PLR is, by definition, above 40 000/µL, which can sometimes be referred to as ‘extreme leukocytosis’.24 Not infrequently, the WBC count can be above 100 000/µL, which is called hyperleukocytosis. The latter is considered a medical emergency as it can lead to increased blood viscosity and tumour lysis syndrome.25 26 Initiation of therapy (hydration, prevention of tumour lysis syndrome, correction of metabolic abnormalities, hydroxyurea, and in selected cases, leukapheresis) should be started as early as possible.26 27
Interestingly, patients with PLR tend to be stable at presentation, in contrast to patients with infection having a similar WBC count.24 If a diagnosis of PLR is established, localising the underlying malignancy should be the next step. Tubal gut endoscopy and radiologic imaging are minimally invasive techniques that can be used.15 Also, since CSTs are metabolically active, they tend to have a high uptake on F-fluorodeoxyglucose positron emission tomography (FDG-PET), which is reported to be effective in localising the underlying malignancy and shortening the diagnostic delay.15–17 21 Due to CSF-induced myeloproliferation, FDG-PET can also show a high and diffuse avidity in the bone marrow, particularly in the axial skeleton.15 17
Some diagnostic criteria have been proposed to help establishing a diagnosis of PLR.14 15 21 These are1 a marked unexplained leukocytosis,2 increased plasma CSF level,3 a decrease in WBC count following therapy causing tumour burden reduction,4 confirmation of G-CSF production in the tumour by immunohistochemistry or immunofluorescence staining, and5 an increase in WBC count or CSF levels if subsequent tumour recurrence/relapse occurs.
Pathological features and differential diagnoses: the role of the pathologist in making the diagnosis
The morphological features of PLR can be identical to features seen in LRs due to other causes, such as infection. They can also be very similar to findings seen in patients being treated with exogenous haematopoietic growth factors (EHGFs), which are commonly used to shorten the period of neutropaenia after myeloablative chemotherapy. Besides causing LR, EHGFs can also lead to a transient increase in peripheral blood blasts count (up to 39% in one study).28 This can mimic acute leukaemia or myelodysplastic syndrome with excess blast. The use of EHGFs is the most common cause of extreme leukocytosis in patients with non-haematolymphoid malignancies, according to a study done on 758 patients.24 In this study, the aetiology of extreme leukocytosis was due to EHGFs in 69% of patients, followed by infection (15%), and lastly PLR (10%). Therefore, before entertaining a diagnosis of PLR, the use of EHGFs must be first excluded.
PLR can present in a similar way to MPNs. Therefore, it is not uncommon for the pathologist to encounter a peripheral blood smear or a bone marrow biopsy from one of these patients. The main differential diagnoses for a LR are as follows: chronic myeloid leukaemia (CML), chronic myelomonocytic leukaemia (CMML) and chronic neutrophilic leukaemia (CNL).3 CNL is much rarer than CML or CMML. Distinguishing the LR from a MPN is the most essential step in reaching the correct diagnosis. The role of the pathologist goes beyond reporting the absence of a haematological malignancy. In addition, he or she must alert the treating physician by pointing out to the possibility of an occult CST.
Peripheral blood findings
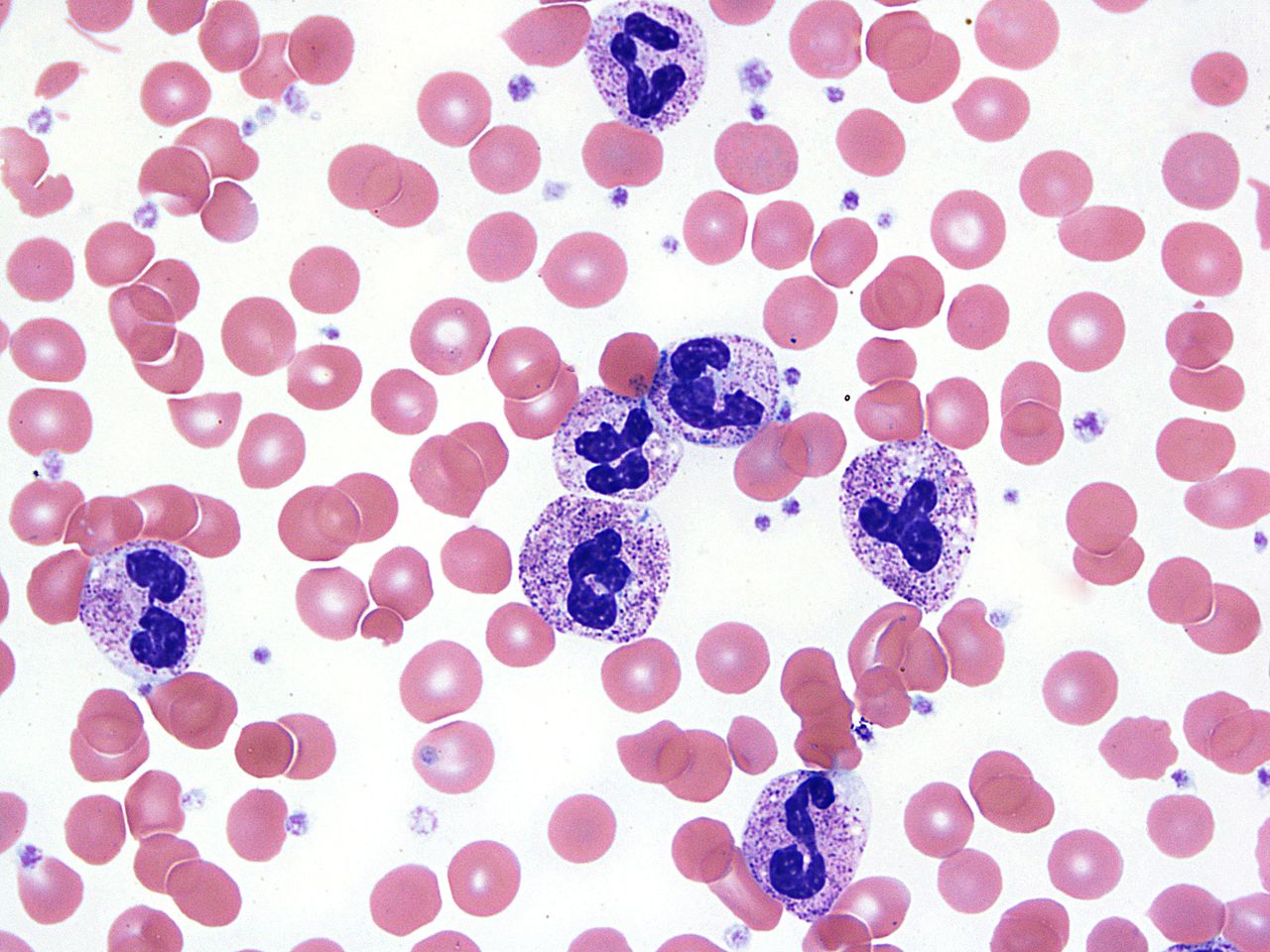
Most PLRs are neutrophilic predominant,24 although eosinophilic PLRs or mixed neutrophilic–eosinophilic PLRs have been also reported.27 29 Blood smears from patients with PLR (figures 1 and 2) show neutrophilia with marked left shift due to increased late granulocytic precursors (band forms, metamyelocytes and myelocytes).3 25 Characteristic features of reactive granulocytosis include toxic granulation, prominent Döhle bodies and cytoplasmic vacuolation.3 23

PLR due to CST showing peripheral blood showing prominent neutrophilia with left shift (bandemia). CST, cytokine-secreting tumour; PLR, paraneoplastic leukemoid reaction.

Neutrophils showing toxic granulation, Döhle bodies and cytoplasmic vacuolations.
Distinguishing LR from CML
In contrast to LR, CML tends to have a characteristic myelocyte peak. The absence of earlier granulocytic precursors (promyelocytes, blast), basophilia or monocytosis can be very helpful in favouring a diagnosis of LR over CML or CMML. In addition, the features of reactive granulocytosis (described above) are less frequently seen in CML and CMML.3 23 Historically, a leukocyte alkaline phosphatase (LAP) score used to be a helpful tool in this setting. The LAP score tends to be low in CML and high in PLR and CNL.3 However, with the widely available BCR/ABL1 assays that can be done on peripheral blood, the LAP score is rarely used nowadays.
Distinguishing LR from CNL
In contrast to LRs, the neutrophilia in CNL is characterised by the lack of left shift. Segmented neutrophils constitute most of the cells. The more immature granulocytic precursors and nucleated erythrocytes are infrequent to find. However, it is important to mention that features of reactive granulocytosis can infrequently be seen in CNL, making the distinction challenging.3 The morphological overlap between PLR and CNL can likely be explained by the mechanistic similarity of these two processes. G-CSF is the ligand for colony-stimulating factor three receptor (CSF3R). In CNL, mutations in the CSF3R gene lead to constitutive activation of downstream signalling protein mediators, leading to granulocytic proliferation.30 In PLR, the ligand (G-CSF) is markedly increased which will lead to a similar end result.
Bone marrow examination and ancillary testing
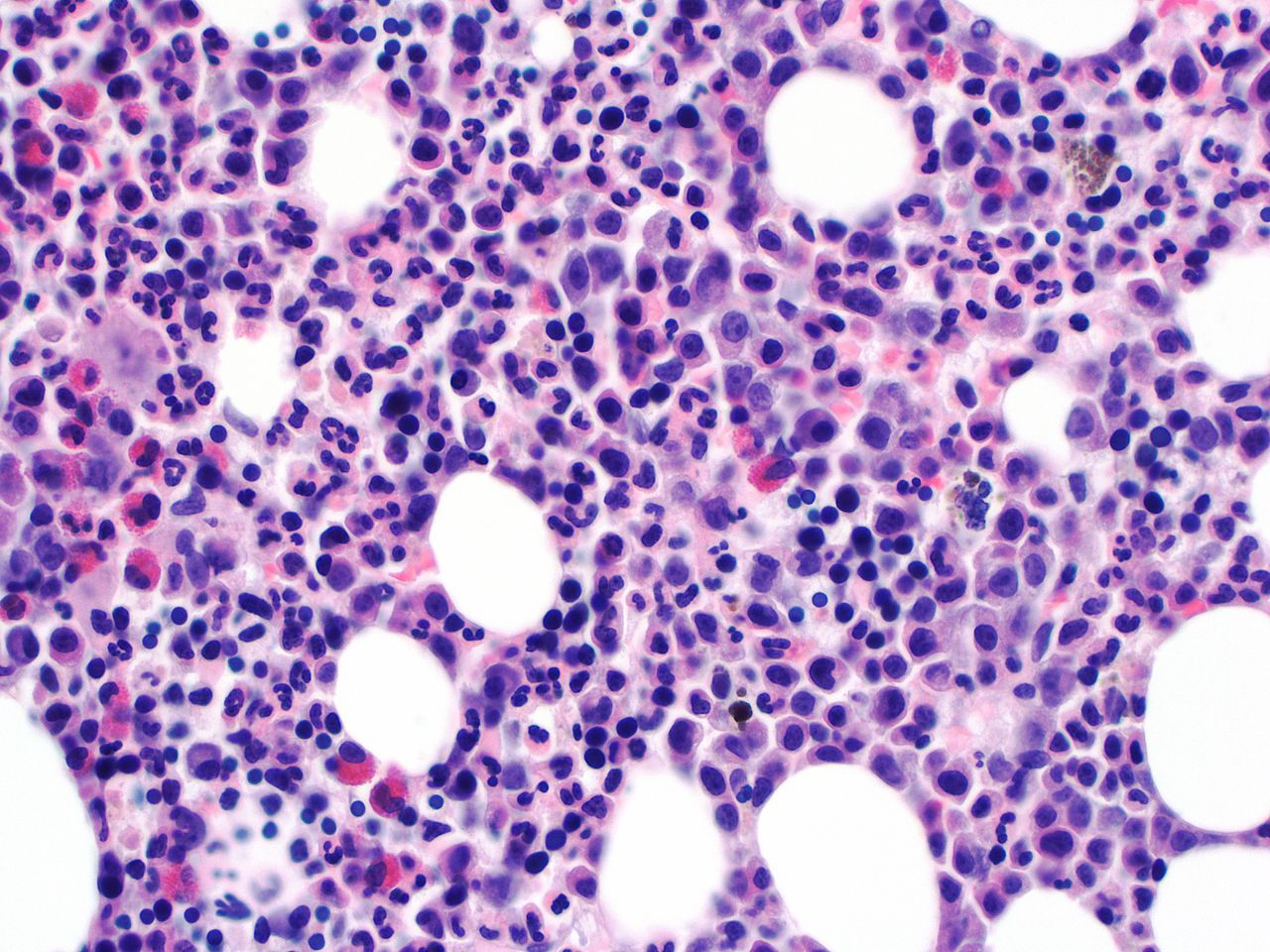
Marrow examination in patients with PLR usually shows hypercellular marrow with myeloid hyperplasia, and no increase in blasts percentage. Left shifted myelopoiesis with toxic granulation can be also seen (figures 3 and 4).18 21 Erythroid precursors and megakaryocytes are usually unremarkable. The characteristic ‘dwarf’ megacaryocytes of CML are not seen. Cytogenetic and molecular studies must be negative for the known molecular drivers of MPNs (BCR/ABL1, CSF3R, TET2, SRSF2, SETPB1 and ASXL1).

Bone marrow aspirate examination showing markedly increased M:E ratio due to increased and left shifted granulopoiesis.

Bone marrow biopsy examination showing markedly hypercellular marrow with increased and left shifted granulopoiesis.
Cytokine blood level
Testing for the suspected cytokine plasma level (G-CSF, GM-CSF, ILs or TNF-α) can be accomplished through enzyme immunoassays or enzyme-linked immunosorbent assays.2 However, these tests are not offered in many institutions.
Histological examination of the CST
Based on our knowledge, all reported PLRs occurred in the setting of malignant tumours. CSTs tend to have a higher grade and a lesser degree of differentiation than other tumours. Most reported tumours are poorly differentiated or undifferentiated.1 6 8–10 12 14 15 21 23 31 Dense intra-tumoural neutrophilic infiltration is a characteristic morphologic finding (figure 5).7 8 CST cells show diffuse cytoplasmic staining with immunohistochemical or immunofluorescence staining with G-CSF or GM-CSF antibody.9 10 14 16 27 However, some CSTs can stain negative even if their CSF-secreting ability has been confirmed by elevated CSF plasma levels.21

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histological examination of the CST showing undifferentiated carcinoma with marked intratumoural neutrophilic infiltration. CST, cytokine-secreting tumour.
Prognosis and treatment
Treating the underlying malignancy is the main therapy for PLR. Surgical resection, radiotherapy and chemotherapeutic agents have been shown to be effective in decreasing WBC counts for responsive tumours.4 10 15 23 However, patients who develop PLR due to CST are generally associated with a worse prognosis and shortened survival period.2 24 They tend to have a large disease burden, either due to a bulky primary tumour or widely metastatic disease. In one study, 76% of patients who had PLR died within 3 months of presentation.24 The previously mentioned mechanisms logically explain the aggressive behaviour of these tumours and the poor prognosis associated with them. The rapid tumour growth and the progressive inflammatory status worsen the general condition of the host. The anti-apoptotic effects, induction of tumour angiogenesis and the suppression of host immunity play an integral role in the aggressive behaviours of these tumours and their ability to metastasize to very rare distant sites (such as the heart or the choroid of the eye).12 13
Conclusion
CSTs causing PLR are highly aggressive tumours associated with a poor prognosis and shortened survival due to multiple mechanisms. Clinicians should keep a high level of suspicion for PLR when dealing with patients having unexplained leukocytosis. Pathologists play an important role in recognising this entity, distinguishing it from a MPN, and guiding the clinical team towards to the possibility of an occult CST. Lack of knowledge of this entity may lead to dramatic negative effects on the patient, including unnecessary diagnostic testing and treatments. When dealing with unexplained leukocytosis, PLR is usually a diagnosis of exclusion, yet the diagnosis should not be delayed as treating the underlying malignancy is the mainstay of treatment.
Take home messages
Paraneoplastic leukemoid reactions are caused by highly aggressive cytokine-secreting tumours that are associated with a poor prognosis. A high level of suspicion for this clinically important entity should be kept when dealing with patients having unexplained leukocytosis.
Myeloproliferative neoplasms are the main differential diagnosis. Therefore, pathologists play an important role in guiding their clinical colleagues towards to the possibility of an occult cytokine secreting tumour.
Lack of knowledge of this entity may lead to dramatic negative effects on the patient, including unnecessary diagnostic testing and treatments.
References
Footnotes
Handling editor Mary Frances McMullin.
Contributors All authors contributed equally to this work.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.