Article Text

Abstract
There is growing emphasis on the potential significance of the placental microbiome and microbiome–metabolite interactions in immune responses and subsequent pregnancy outcome, especially in relation to preterm birth (PTB). This review discusses in detail the pathomechanisms of placental inflammatory responses and the resultant maternal–fetal allograft rejection in both microbial-induced and sterile conditions. It also highlights some potential placental-associated predictive markers of PTB for future investigation. The existence of a placental microbiome remains debatable. Therefore, an overview of our current understanding of the state and role of the placental microbiome (if it exists) and metabolome in human pregnancy is also provided. We critical evaluate the evidence for a placental microbiome, discuss its functional capacity through the elaborated metabolic products and also describe the consequent and more established fetomaternal inflammatory responses that stimulate the pathway to preterm premature rupture of membranes, preterm labour and spontaneous PTB.
- maternal-fetal
- placenta
- pregnancy
- microbial pathogenic
- immunopathology
Statistics from Altmetric.com
Introduction
This review provides an overview of our current understanding of the state and role of the not-yet proven placental microbiome and metabolome in human pregnancy. The human microbiome has existed as a complex, symbiotic ecosystem of microorganisms inhabiting human mucosal tissues and skin, yet its importance in health and disease has only been reported in recent years.1 Growing evidence suggests a vital role for the human microbiome in immunity, metabolism and cancer but clarity remains required regarding whether microbiome dysregulation is the cause or consequence of these processes and conditions.2–5 Dysbiosis of the gut microbiome has been implicated in influencing tumourigenesis and cancer progression in gastrointestinal and systemic cancers6 as well as in the development of polycystic ovary syndrome,7 type 2 diabetes8 and even mental health disorders such as anxiety and depression.9 Research suggests a possible contribution from a deranged maternal gut microbiota in the development of atopy, the genetic tendency to develop allergic diseases such as rhinitis and asthma and autoimmune phenotypes in the neonate,4 along with an increased risk of necrotising enterocolitis and sepsis.10 Furthermore, emerging evidence of the interactions between placental and fetal gut and lung microbiomes highlights that dysregulation of these fetal microbiomes related to placental dysbiosis may influence short-term and long-term neonatal complications such as bronchopulmonary dysplasia and necrotising enterocolitis among other morbidities.10 Nonetheless, whether the maternal gut microbiota, and the subsequent impact on metabolic, hormonal and immunological factors during gestation, exerts any influence on the pregnancy itself remains unclear.11
However, there is consensus on the role of the vaginal microbiome in health with a dominance of protective Lactobacillus species, which make up >70% of resident bacteria, found in the vagina of healthy, reproductive-age women.11–14 Lowered disease risk and increased protection from opportunistic pathogens is considered to be the benefits of this dominance and the low pH of 4.5 observed in humans. Additionally, decreased relative abundance of Lactobacillus species in the vagina is strongly linked to bacterial vaginosis (BV) resulting in increased anaerobic bacterial respiration, pH values ≥4.7 and increased risks of infertility, maternal infection (particularly sexually transmitted infections), preterm premature rupture of membranes (PPROM) and preterm labour (PTL) through microbial ascent to the uterus.12–15
The two-part Human Microbiome Project (HMP), spanning almost 15 years, has sought to answer questions surrounding the uniqueness of the microbiome between humans and throughout different life stages, as well as how diversity contributes to function and dysfunction.15 16 The first part of the HMP revealed a microbiome distinctly different within each individual’s niche in addition to a ‘personalised’ microbiome with discrete variants. The second part determined how changes to the host–microbiome interactions affect the stressors for three conditions—pre-diabetes, inflammatory bowel diseases and pregnancy including preterm birth (PTB). Lactobacillus dominance of the vaginal microbiome positively correlated to term birth. A ‘signature’ microbiome was described for a high risk of PTB. However, pregnancy-related microbiomes for other tissues such as the placenta, uterus, breast milk or blood were not explored.15
Despite this apparent oversight, evidence showed support for the translocation of maternal gut bacteria to breast milk and other extraintestinal sites during pregnancy.4 They also showed that the maternal gut microbiome exerts an influence on neonatal immunity most notably through colonisation of the neonatal gut during vaginal delivery.4 17 18 Contrastingly, reports of haematogenous and in utero transmission of bacteria to identifiable placental and amniotic fluid microbiomes challenge the decades-old ‘sterile womb’ paradigm.19–23
Given the above raging controversy, this review discusses in detail the pathomechanism of placental inflammatory responses (PIRs) and the plausibly resultant maternal–fetal allograft rejection in both microbial-induced and sterile conditions. We also highlight some potential placental-associated predictive markers of PTB for future investigation. The evidence for and against a placental microbiome is described and how this may interact with the placental metabolome during human pregnancy is also provided in the context of the more established fetomaternal inflammatory responses that stimulate the pathway to PPROM, PTL and spontaneous PTB.
Placental inflammatory response
Histological features
Spontaneous preterm birth is often associated with ascending intrauterine infection that triggers both maternal and fetal inflammatory responses (FIRs), collectively referred to as the PIR. PIR is associated with poor neonatal outcomes including low birth weight, sepsis, intraventricular haemorrhage, cerebral palsy, bronchopulmonary dysplasia and neonatal mortality.24 25 The inflammatory responses of the placenta reflect the maternal and FIRs. PIR begins as a maternal inflammatory response with subsequent FIRs elicited if acute chorioamnionitis persists and increases in severity.24
The inflammatory response of the placenta that extends into the chorion, amnion or decidua is referred to as maternal inflammatory response. The hallmark of this response is histological chorioamnionitis (HCA). On the other hand, inflammation that infiltrates the chorionic plate, umbilical cord (funisitis) and fetal blood vessels (vasculitis) is regarded as FIR, which is defined clinically as a fetal cord blood plasma interleukin (IL)-6 concentration >11 pg/mL,24 and frequently associated with microbial invasion of the amniotic cavity.24 26
The fetus’ capacity to respond to bacterial colonisation develops after 20–21 week’s gestation and manifests as inflammation of the umbilical cord (funisitis). This fetal response begins in the umbilical vein (phlebitis) before progressing to one or both umbilical arteries (arteritis) and into the Wharton’s jelly, with this progression being a good pathological indicator of the stage and severity of the FIR.27 28 Some researchers regard both funisitis and chorioamnionitis as hallmarks of fetal inflammatory response syndrome (FIRS); however, the FIR indicates an increased severity and advancement of acute chorioamnionitis.26 29 Both maternal and FIRs are associated with PPROM but FIR in the placenta is usually more severe than MIR and more frequently associated with poor neonatal outcome.25 30 Preterm parturition is usually associated with HCA and infants born in such situation are at greater risk of sepsis and poor neurological, cardiorespiratory, gastrointestinal (including necrotising enterocolitis) and renal outcomes.24 25 31 32
There is often chorioamnionitis (inflammation in the chorion and amnion) at placental examination with associated high concentration of cytokines in the amniotic fluid.33 However, signs of intra-amniotic inflammation have been observed without elevated amniotic fluid cytokine levels.33 Early detection has resulted in the identification of several inflammatory markers associated with PIR.
Spontaneous PTB between 24 and 32 weeks are less common (<2% of all births) than late preterm births but are associated with greater risk of morbidity and mortality and evidence of intrauterine infection and/or inflammation.34 HCA is defined by inflammation of the chorion, amnion and placenta.31 It is a frequent observation (~70%) in PTBs between 20 and 24 weeks gestation, but rare (16%) in deliveries after 34 weeks.26 HCA is observed in 10% of term births, 30% of PTL with intact membranes and 50% of PPROM, with the highest frequency of 80%–90% in miscarriages at 20–24 weeks.35 Despite the classic observation of HCA and high amniotic fluid concentrations of chemocytokines or matrix degrading enzymes in intrauterine infection and/or inflammation, only a few of such pregnancies (5%–12% at term; and 20% preterm with preterm rupture of membranes (PROM))36 exhibit clinical signs of chorioamnionitis.34 HCA is about three times more common than clinical chorioamnionitis, which is characterised by maternal fever, tachycardia, uterine tenderness, leucocytosis and PROM31; and diagnosed by positive amniotic fluid culture of bacteria such as genital mycoplasmas.37 Intrauterine infection may also occur through haematogenous spread, by invasive medical procedures, or by retrograde passage from the peritoneal cavity through the Fallopian tubes, but most commonly by ascent from the vagina associated with a dysbiotic microbiota.34 37
Immune profile
The placental microenvironment in healthy pregnancies is biased toward a T-helper (Th)2 (predominantly anti-inflammatory, eg, IL-4 and IL-10) cytokine milieu,38 and reversal to Th1 cytokine (eg, tumour necrosis factor α, interferon-gamma (IFN-γ), IL-2 and IL-1β) dominance is associated with labour.39 The balance between Th1 (cell mediated and proinflammatory) and Th2 cytokines is considered a measure of the immune environment.40 Alteration in this cytokine balance can lead to a breakdown of the maternal–fetal tolerance and rejection of the semiallogenic fetus.41 Increased expression of proinflammatory cytokines in the placenta, cord blood, maternal blood and cervicovaginal fluid is associated with poor pregnancy outcomes including placental dysfunction, intrauterine growth restriction (IUGR), preeclampsia and PTB.38 40 42 43 Placentas from women with PPROM and PTB indicate a bias towards Th1 cytokines IFN-γ, IL-2 and IL-12 compared with term placentas that show higher IL-4 and IL-10 (Th2) levels.39 Elevated levels of IL-6 (produced by Th2 cells) and IL-8 (a chemokine produced by macrophages and monocytes) are also observed in preterm compared with term placentas. IL-6 is a highly sensitive and specific indicator of infection-induced PTL, while up to a 1000-fold increase in IL-8 expression have been observed in PTL with evidence of chorioamnionitis.39
Choriodecidual colonisation by bacteria with release of pathogen-associated molecular patterns (endotoxins and exotoxins) activate pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) and nod-like receptors that are expressed by amnion epithelial cells, intermediate trophoblasts in the chorion, decidual cells, macrophages and neutrophils.44 These PRRs activate the pleiotropic transcription factor nuclear factor-κB (NF-κB) signal transduction pathway. NF-κB regulates the expression of proinflammatory cytokines, chemokines and antimicrobial peptides (eg, defensins).34 37 45 HCA in the setting of PTB is associated with significantly increased chorionic expression of TLR-1 and TLR-2 compared with PTB without HCA.35 Regardless of membrane status (intact or ruptured) and gestational age (term or preterm), HCA is associated with increased expression of TLR-2 and TLR-4 on chorioamniotic membranes.45 TLR-2 recognises microbial products of gram-positive bacteria (peptidoglycan, lipoteichoic acid), genital mycoplasmas and fungi, while TLR-4 recognises bacterial endotoxin (lipopolysaccharide).45 TLR-4 signalling pathway activates NF-κB through the recruitment of adapter proteins (MyD88, IRAK1/4 and TRAF6) and activation of intermediate kinases (RIP1, TAB2/3, TAK1 and IKKα/β; figure 1).34 Other signalling pathways (p38MAP kinase/CREB) and modulators (histone deacetylase) that interact unselectively with NF-κB signalling are also involved.34 46 Key regulators of allogenic tolerance are decidual macrophages, specifically of the M2 anti-inflammatory phenotype, and regulatory T cells (Tregs), which exhibit immunosuppressive properties. The placenta is capable of inducing these homeostatic macrophages and regulatory T cells to establish and maintain pregnancy with accumulation of Tregs having been observed in the decidua.47

Microbial and sterile inflammatory signalling pathways associated with preterm labour and birth. Both pathogen-associated and damage-associated molecular patterns (PAMPs/DAMPs) activate nuclear factor kappa B (NF-κB) by interacting with pattern recognition receptors (toll-like receptor (TLR) and nod-like receptor (NLR)) on immune cells and gestational tissues. The pleiotropic NF-κB regulates the expression of proinflammatory mediators that activate the tripartite common pathway of labour and if before 37 completed weeks of gestation, preterm birth (PTB). CCL2, chemokine (C-C motif) ligand; cffDNA, cell-free fetal DNA; GCSF, granulocyte colony-stimulating factor; HMGB1, high-mobility group box 1; Hsp70, heat shock protein 70; IFN-γ: interferon-gamma; IKKα/β, inhibitor of nuclear factor kappa B kinase subunit alpha/beta; IκB, inhibitor of nuclear factor of kappa B; IL-1α, interleukin-1alpha; IL-1β, interleukin-1-beta; IL-6, interleukin-6; IL-8, interleukin-8; IRAK1/4, interleukin-1 receptor-associated kinase ¼; LPS, lipopolysaccharide; LTA, lipoteichoic acids; MMP, matrix metalloproteinases; MSU, monosodium urate; mtDNA, mitochondrial DNA; MyD88, myeloid differentiation primary response 88; PG, prostaglandins; PGN, peptidoglycans; RIP1, receptor interacting protein 1; TAB2/3, transforming growth factor beta-activated kinase 1-binding protein 2/3; TAK1, transforming growth factor beta-activated kinase 1; TRAF6, tumour necrosis factor receptor-associated factor.
Cytokine profile
The downstream effect of NF-κB activation is the release of proinflammatory chemocytokines, which induce neutrophil activity, synthesis and release of prostaglandins (PG), contractile associated proteins, and matrix metalloproteinases (MMPs) in the placental tissues and fetal membranes. The ultimate effect is increased myometrial contractility, cervical remodelling, membrane rupture and PTB (figure 1),37 46 which can occur due to premature or pathological activation of NF-κB.48
On the other hand, IL-10 inhibits the activity of NF-κB,49 and downregulates Th1-associated cytokines and PGE2 expression.50 IL-10 expression is stimulated by bacterial colonisation of the fetal membranes and amniotic cavity, and inflammation-associated intracellular signalling pathways, for example, MyD88, p38MAP kinase, CREB and STAT.46 Levels of IL-10 and IL-4 are elevated in placentas of women who delivered at term,51 while placental IL-10 expression is reduced in pregnancies complicated by chorioamnionitis and PTL.52 Similarly, low levels of IL-10 have been observed in umbilical cord blood of preterm neonates. However, high plasma levels of IL-10 have been reported in women who experienced PTB.53
Another anti-inflammatory cytokine, transforming growth factor-β2 (TGF-β2) is also secreted by the placenta.51 Low cord blood levels of TGF-β but elevated IL-10 have been found in women with placental inflammation.54 High levels of TGF-β and IL-4 in maternal plasma have also been associated with high risk of PTB <35 weeks with HCA.40 In a recent case-control study, low serum levels of IL-10 or TGF-β or both simultaneously between 22 and 25 weeks’ gestation were associated with high risk of PTB independent of the presence of BV.55 These regulatory cytokines55 prevent inflammation-mediated damage to the fetus.51
Another endogenous protein associated with immune reactivity and homeostasis in pregnancy is antisecretory factor (AF). AF is present in the placenta and other human tissues and body fluid. It is also expressed by macrophages and dendritic cells with potent anti-inflammatory effects. It usually exists in an inactive form and becomes activated in response to exposure to bacterial toxins as part of the innate immune response. Placenta levels of active AF are inversely related to inflammation and decrease in PTB. It has been suggested that AF regulates placental inflammation during gestation by converting complement factor 3 to its inactive form and stimulating the activity of proteasomes.56
Bacterial colonisation of the placenta in the latter half of pregnancy elicits the so-called FIRS, through the stimulation of the fetal hypothalamic pituitary axis and the production of corticotropin-releasing hormone (CRH) from the placenta. Subsequently, there is increased fetal cortisol biosynthesis and decreased maternal cortisol metabolism. Cortisol stimulates PG synthesis and through a positive feedback loop induces placental CRH production. PG production is enhanced by concomitant downregulation of PG dehydrogenase within the chorion and the production of PTGS-2. Both CRH and PGs stimulate MMP production and infiltration of inflammatory mediators.37 44
Sterile inflammation
Some women with signs of intra-amniotic inflammation, for example, elevated amniotic fluid cytokines do not demonstrate clinical intra-amniotic infection.33 For instance, in the absence of positive amniotic fluid bacterial cultures, elevated amniotic fluid IL-6, IL-1α, IL-1β levels have been shown to correlate with PTB before 34 weeks.57 Elevated placental58 59 and cervical50 cytokine levels have been described in women who delivered preterm without histologically confirmed intrauterine infection. Spontaneous PTB without infection is more common than PTL associated with infection.46 60 However, some pregnancies without microbial invasion of the amniotic cavity but with bacteria localised to the fetal membranes may be misclassified as ‘sterile’. More so, because antibiotics are generally ineffective in preventing PTL and birth in women with infection, such PTBs may be the result of inflammation rather than infection.34 Among other factors, the degree of immune response mounted by the host against the infectious agent is crucial to the ultimate reproductive outcome. This is because a significant proportion of women with inflammation of the gestational tissues who deliver prematurely do not have infection.46 61 Hence, for more effective prevention/treatment of PTB, coadministration of antibiotics and anti-inflammatory agents has been recommended.34
Sterile inflammation may be induced by cellular stress, tissue damage and danger signals and is mediated by damage-associated molecular patterns or alarmins, which activate PRRs. The non-microbial molecular drivers of sterile inflammation are usually associated with cell damage or death and are either intracellular—high-mobility group box 1, cell-free DNA, uric acid, heat shock proteins (eg, Hsp70), IL-1α and mitochondrial components (DNA, ATP, N-formulated peptides); or extracellular—extracellular matrix proteins (fibronectin, hyaluronan), oxidised phospholipids, fatty acids, advanced glycosylation end products, lipid oxidation products and oxysterols. These alarmins are released by the placenta and fetal membranes and provoke inflammation by activating PRRs on gestational tissues resulting in activation of NF-κB and p38MAP kinase signalling pathways which stimulate transcription of inflammatory genes. The ultimate outcomes include placental dysfunction, IUGR, preeclampsia and PTB (figure 1).42 46 A detailed review of the role of cytokines in PTL is provided by Farina and Winkelman.50
Chronic versus acute placental inflammation
Placental infections affect different histological components of this complex organ and are termed chorioamnionitis, intervillositis, villitis and deciduitis. Depending on the pathological features expressed, placentitis could be acute or chronic. Both acute and chronic infectious villitis result from direct haematogenous spread of pathogens to the placenta, though whether the inflammatory cell types are neutrophils or macrophages and lymphocytes defines an acute or chronic villitis, respectively.29 62 Where cultures, PCR or additional methods fail to demonstrate an infection, a diagnosis of villitis of unknown aetiology (VUE) is made, a condition speculated to be related to dysregulation of the maternal immune system.63 The prevalence of VUE varies between studies with a range identified between 2% and 33.8% of all placentas having a positive diagnosis; it has been suggested that differences in diagnostic criteria and sampling protocols may account for this broad range. VUE is mainly associated with term placentas and is often regarded as being a benign placental inflammatory lesion despite the more severe grades being associated with adverse neonatal outcomes.29 63
An infiltration of neutrophils characterises acute infections, either chorioamnionitis or villitis, whereas the hallmark of chronic chorioamnionitis (CCA) is an infiltration of lymphocytes to the chorionic plate and chorionic membranes. Acute chorioamnionitis presents with a thickened amniotic membrane which can be easily separated from the amnion due to adhesions between the layers,62 64 a feature which distinguishes it from the rarely observed condition, CCA.62 64 VUE is diagnosed where there is no evidence to support an infectious aetiology and an infiltration of Hofbauer cells (fetal macrophages) and maternal T-lymphocytes are demonstrated with histological staining techniques. Uniquely, the inflammatory processes in VUE involves immune cells from two different hosts and shows maternal cells damaging fetal tissue and a response resembling allograft rejection.29 63 65
Due to the controversy of terminologies used in describing placental lesions and diagnostic criteria, a group consensus has been developed to reduce definition and sampling differences between laboratories and to standardise grading and staging of inflammation, as detailed in table 1.30
Amsterdam placental workshop group consensus on staging and grading of maternal and fetal inflammatory responses
Acute inflammatory response to ascending infection is more common than chronic inflammatory lesions during gestation. Acute inflammatory response is characterised clinically and pathologically as fetomaternal neutrophilic infiltrates (table 2). Neutrophilic migration to the chorioamnion is induced by elevated concentrations of intra-amniotic neutrophil chemokines such as IL-8.29 Increase in histological severity in both maternal and fetal acute inflammatory response usually correlates with time of onset of ascending infection during gestation. Usually, maternal response precedes the fetal response.66
Characteristics of acute placental inflammatory responses (Amsterdam criteria30)
On the other hand, the less well understood chronic inflammatory response could be focal or multifocal but without obvious relationship between aetiology or clinical outcome and severity of inflammation. Due to the expansive collection of placental locations at which chronic inflammatory cells can be observed, characterising chronic inflammatory lesions is often challenging. CCA features in about 39% and 34% of cases of PPROM and PTB, respectively.29 It is especially common in late PTB which constitute up to 70% of premature deliveries. Additionally, CCA and elevated intra-amniotic CXCL10 (T-cell chemokine) have been demonstrated in up to 60% of cases of fetal death suggesting a role of maternal antifetal rejection in certain cases of idiopathic fetal death.29 Where CCA is observed histologically, a concomitant chronic deciduitis or VUE is commonly diagnosed. Similarly to VUE, CCA is regarded as being immunological in origin, though it is associated mainly with preterm births rather than term deliveries.64
Chronic inflammatory response is characterised by incidental microscopic identification of chronic inflammatory cells including lymphocytes, plasma cells, histiocytes (tissue macrophages) and eosinophilic infiltrates, most commonly observed in the choriodecidual border of the chorion laeve (table 3).64 The degree and severity of T-lymphocyte infiltration are usually not up to the neutrophilic infiltration observed in acute chorioamnionitis. A ratio of neutrophil to lymphocyte shows useful predictive accuracy for PIR with 71.4% sensitivity, 77.9% specificity, 80.7% positive predictive value and 67.8% negative predictive value.24
Characteristics of chronic placental inflammatory responses (Amsterdam criteria30)
It is proposed that the migration of CXCR3+ T cells from the maternal (decidua) to fetal tissue (chorioamniotic membranes) (amniotropism) is induced by high concentration of amniotic fluid CXCL10 and overexpression of CXCL9, CXCL10 and CXCL11 in the chorioamnion (figure 2).29 This is supported by the observation of elevated amniotic fluid CXCR3 and its ligands CXCL9 and CXCL10 in women who experience PTL and PTB, and lesions of maternal antifetal rejection (eg, CCA, chronic histiocytic intervillositis, VUE and chronic deciduitis with plasma cells) compared with those without such lesions even though they deliver preterm. CXCR3, CXCL9 and CXCL10 are also higher in women with spontaneous miscarriage or PTL (sPTL) and PTB compared with those with labour at term. In addition, irrespective of the presence or absence of placental lesions, elevated CXCR3 is associated with PTL compared with labour at term. Interaction of CXCR3 and its IFN-γ-inducible ligands (CXCL9, CXCL10 and CXCL11) stimulates migration of maternal CD8 cytotoxic T-cells from peripheral circulation to the decidua and chorioamnion, increase production of Th1 and reduce synthesis of Th2 cytokines (figure 2). However, this phenotype is not observed in physiological labour at term.67 The blockage of CXCR3 could attenuate the chronic inflammatory response associated with PTL and this could be beneficial in preventing maternal antifetal rejection and prematurity.

Disruption of maternal–fetal tolerance during pregnancy. Chronic inflammatory lesions of the placenta are associated with overexpression of CXCR3 (a transmembrane G-protein coupled receptor) by the villous cytotrophoblasts and syncytiotrophoblasts, chorioamnion and decidua. Binding of CXCR3 to its interferon γ (IFN-γ)-inducible chemokine ligands (CXCL9, CXCL10 and CXCL11) generates a T-cell chemokine gradient from the amniotic fluid and increases CD8+ cytotoxic T cell infiltration of the decidua and fetal membranes. This stimulates cell-mediated and antibody-mediated immune responses that lead to chronic chorioamnionitis, villitis of unknown aetiology and chronic deciduitis with plasma cells that are placental lesions associated with maternal antifetal rejection, fetal growth restriction and spontaneous preterm labour (sPTL).29 67.
Loss of allogenic tolerance is another potential mechanism for PTB. This occurs via an imbalance in Tregs, inflammatory Th17 cells and/or decidual macrophages. Tregs are essential for controlling excessive immune responses and tolerance and are influenced by resident microbiota in tissues other than the placenta, such as the gut, oral mucosa and the vagina. Induction and maintenance of Tregs rely on the local microbiota and changes to the Tregs/Th17 balance or microbial dysbiosis are linked to infections, increased neutrophil infiltration and a breakdown in commensal tolerance in mucosa. It is therefore not unrealistic for the placenta to exhibit a similar response whereby dysbiotic microbiota results in maternal antifetal rejection.47 68
CCA, chronic histiocytic intervillositis, VUE and chronic deciduitis each present individual inflammatory features; however, they are rarely observed in isolation. Most commonly two or more of these conditions are seen and are termed overlap chronic placental inflammation (oCPI), which presents with unique inflammatory features separate to those noted individually.
There is also the hypothesis that oCPI, that is, the presence of CCA, chronic histiocytic intervillositis, VUE and chronic deciduitis together in a tissue, could show unique inflammatory features compared with those seen in the rare cases when these conditions occur individually. For example, oCPI may represent an IgA-mediated immunological response characterised by elevated expression of CXCL13,69 which is selectively chemotactic for B cells through its interaction with CXCR5.70 71 This hypothesis is derived from the inclusion of IgA positive plasma cells by oCPI-associated chronic deciduitis compared with isolated chronic deciduitis.69
The eosinophils contribute to the fetal inflammatory infiltrate in both acute chronic vasculitis and eosinophilic/T-cell chronic vasculitis (table 3).66 Many cases of idiopathic chronic placental inflammation, for example, CCA or VUE are reflective of fetal allograft rejection characterised by infiltration of maternal cytotoxic lymphocytes (CD8+ T cells) and presence of fetal human leucocyte antigen-specific antibodies in maternal serum.29 56
Furthermore, amniotic fluid concentration of glycodelin-A, which is associated with the maintenance of maternal tolerance against the semiallogenic placenta/fetus, is decreased in CCA. This is in addition to 31 differentially expressed proteins in cases of CCA compared with cases of acute chorioamnionitis or controls without inflammatory lesions.29
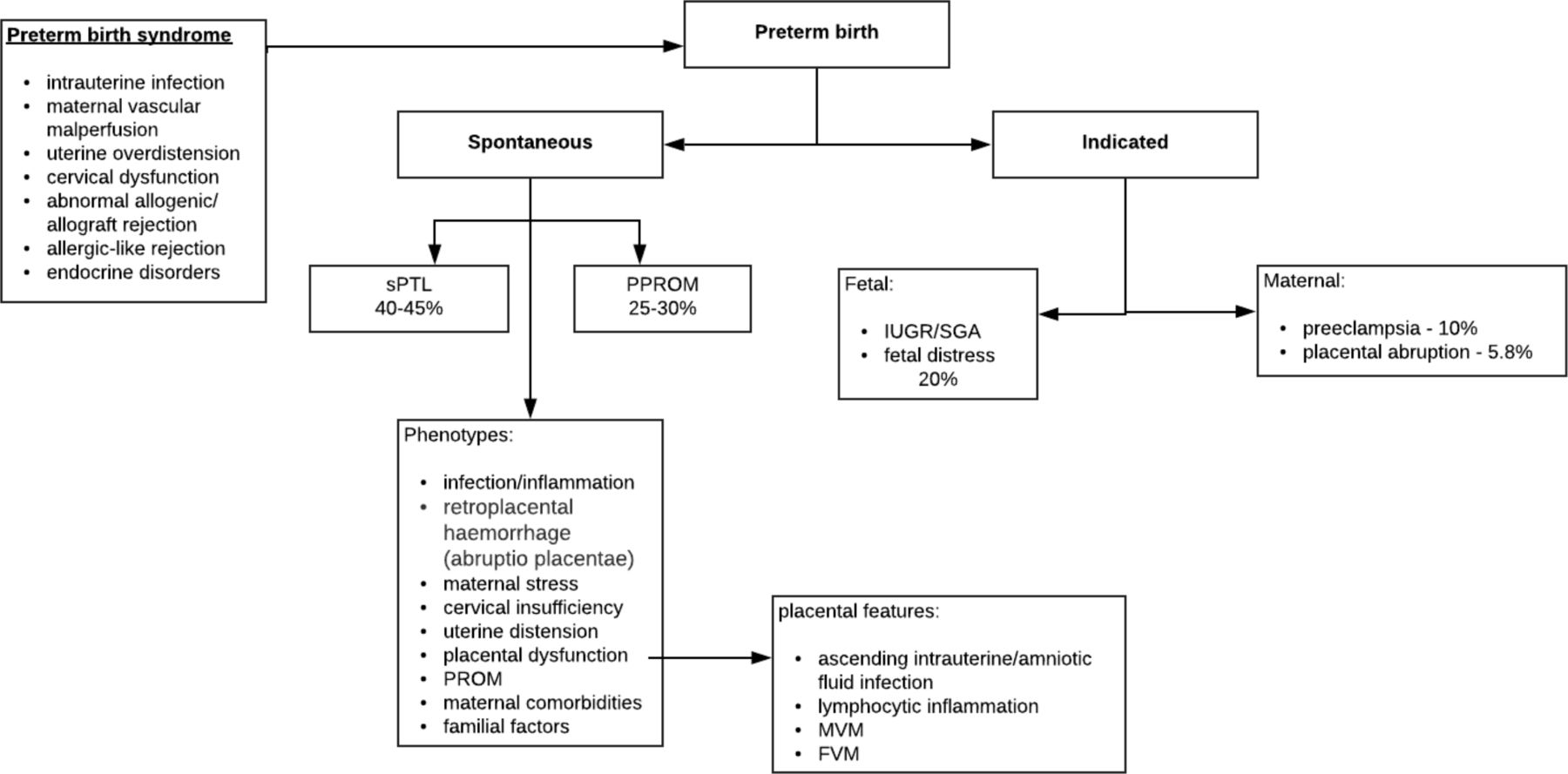
The clinical presentation of PTB (especially <34 weeks’ gestation) is associated with placental pathological features (figure 3). Ascending intrauterine infection/amniotic fluid infection has been shown to be associated with maternal vascular malperfusion (MVM) and common in cases of PPROM. Women with preeclampsia show changes associated with severe MVM. Irrespective of the presence of intrauterine/amniotic infection, lymphocyte inflammation (CCA and VUE) was identified in all clinical presentations of PTB implicating maternal antifetal immunological rejection. CCA and basal villitis are also associated with cases of placental abruption showing laminar decidual necrosis and acute retroplacental haemorrhage.72

{kind=link}
{kind=link}
{kind=link}
Association of clinical presentation of preterm birth and placental pathological features. Lymphocytic inflammation manifests as chronic chorioamnionitis, villitis of unknown aetiology and chronic deciduitis related to preeclampsia and essential hypertension, all of which are placental inflammatory lesions observed in maternal fetal allograft rejection. Histologically, maternal vascular malperfusion (MVM) correlates with uterine ischaemia.87 FVM, fetal vascular malperfusion; IUGR, intrauterine growth restriction; PPROM, preterm premature rupture of membrane; SGA, small for gestational age; sPTL, spontaneous preterm labour.88–90
Placental microbiome
A successful pregnancy largely depends on an optimal immune response to the fetal allograft and microbial and other stimuli.20 73–75 Interruption of the tolerogenic state associated with a healthy pregnancy results in maternal antifetal rejection, placental damage and pregnancy complications including fetal growth restriction and sPTL. This allograft rejection is either cellular (T-cell) or antibody (humoral) mediated; an extreme form of such rejection is fetal death similar to graft failure in organ transplantation.29 It is hypothesised that VUE, an under-recognised yet significant diagnosis, which is discussed in more detail within this review, results in a distinct fetal systemic inflammatory response akin to allograft rejection or graft versus host disease despite the absence of identifiable pathogens.29 63
Evidence in favour of a placental microbiome
Detection of urogenital microbes in the uterus or bacterial colonisation of the placenta and/or amniotic membranes has traditionally been speculated to result in subclinical infection, such as chorioamnionitis, and a concomitant initiation of labour.20 76–78 Microbes ascending from the vagina, including group B Streptococcus, and Ureaplasma and Mycoplasma species, have been associated with chorioamnionitis, placental and membrane colonisation and PTB. Interestingly, non-urogenital bacterial species, such as Streptococcus and Fusobacterium sp, which are normally found in the oral cavity, have also been linked to placental and fetal colonisation and chorioamnionitis, further supporting the hypothesis of haematogenous transfer of microbial organisms to contribute to a placental microbiome.75 79 80 Placentas delivered by sterile caesarean section following term pregnancy with an absence of infection were found to harbour bacteria, as did amniotic fluid from women who had intact membranes.5 Doyle et al 81 used 16S ribosomal RNA gene sequencing to analyse placental and fetal membrane microbial communities in a rural Malawi cohort reported a placental microbiome at delivery distinct from the vaginal and oral microbiomes. An increased bacterial load of Fusobacterium nucleatum, Ureaplasma sp and Gemella asaccharolytica was identified in the fetal membranes and this was shown to correlate with shorter gestation and PTB.81 A placental microbiome resembling that of the oral cavity, specifically the tongue and tonsils, was identified in a study of 320 women. While of a low abundance, bacteria of the species Firmicutes, Tenericutes, Proteobacteria, Bacteroidetes and Fusobacteria were found to be present in placentas following 16S rRNA sequencing20 challenging previous constructs that the placenta has no microbiome.20 73–75
Evidence against a placental microbiome
Conversely, many sequencing-based studies have failed to identify a placental microbiome,74 82–85 suggesting instead that contamination by reagents used in 16S gene sequencing is the cause of microbial identification and reported placental microbiomes, a recognised vulnerability of sequencing studies, especially when studying tissue with a low bacterial abundance such as the placenta.86 Alternative sources of contamination which may be misinterpreted as suggesting a placental microbiome have been attributed to environmental sources, either during or after delivery or from maternal faeces.20 84–86 The lack of a placental microbiome has garnered further support following a DNA-sequencing approach study of villous tissue samples from 537 women including negative controls of samples free of biological material. This research found no evidence of a functional placental microbiome and demonstrated that bacterial DNA contamination was from laboratory reagents or acquisition during labour or delivery.74
Placental microbial metabolite profile
By combining the application of 16S ribosomal DNA-based and whole-genome shotgun metagenomic sequencing analyses with multiplex bead-based immunoassay, researchers have observed that changes in placental microbiota that influence delivery outcome are accompanied by variations in microbiota-induced metabolic pathways.20 73 75 These alterations appear not to be influenced by whether the placentas were delivered vaginally or through caesarean section.20
The placentas of women who deliver at term without chorioamnionitis show significant positive correlation between Acinetobacter sp and Streptococcus thermophilus, and functional pathways involving metabolism of cofactors and vitamins such as riboflavin, nicotinate and nicotinamide, vitamin B6, pentothenate and CoA. These pathways were similarly related to Lactobacillus crispatus, Acinetobacter sp, Streptococcus sp, Fusobacterium sp and Enterobacter sp in preterm women without chorioamnionitis.75 However, women with chorioamnionitis irrespective of birth outcome showed alterations in biosynthesis of secondary metabolites (eg, phenylpropanoid, stilbenoid, diarylheptanoid and gingerol) and lipid (glycerolipid, glycerophospholipid, arachidonic acid and unsaturated fatty acids) metabolism associated with increased abundance of oral commensal bacteria—Streptococcus thermophilus and Fusobacterium sp Furthermore, in the term women, those with chorioamnionitis had significantly lower butanoate and riboflavin metabolism compared with those without chorioamnionitis. Butyrate from butanoate metabolism inhibits inflammation in the gut,73 while decreased riboflavin metabolism is associated with inflammation. This altered secondary metabolite biosynthesis is most likely associated with the observed histological inflammatory responses in term placentas with chorioamnionitis.75
On the other hand, the preterm women with chorioamnionitis showed significantly increased pentose phosphate pathway (PPP), glycerophospholipid metabolism and biosynthesis of siderophores compared with those without chorioamnionitis. The increased PPP is considered to be related to the reduced amniotic fluid glucose observed in chorioamnionitis and PTB as glucose feeds into the PPP.87 Glycerophospholipids are metabolised to arachidonic acid, which promotes inflammation and PG synthesis, both required for initiation of labour.75 Siderophores are iron-chelating molecules released by aerobic and facultative anaerobic bacteria that require iron for proliferation.88 89 Some of such bacteria (eg, Gardnerella vaginalis) have been associated with dysbiosis and adverse reproductive outcomes,89 compared with lactic acid-producing Lactobacilli that do not require iron for growth and proliferation.89 Increased siderophores in preterm placenta is also possibly related to iron deficiency anaemia. It may also be a causative factor that triggers PTB as response to aggravated iron status.73
Elevated amounts of folic acid has also been observed in the placenta of preterm women without excess gestational weight gain.73 This observation is one of several mixed and contradictory reports present in the literature.73 For instance, one study suggested that folic acid supplementation initiated beyond the first and second trimester is associated with increased risk of PTB but this was reported without accounting for confounding factors that may affect this association.90 This observation conflicts with the slight reduction in PTB risk observed in other studies,91 highlighting the need for further studies to confirm the role of folic acid supplementation before and/or during pregnancy.90 91
Conclusion and future directions
There is a growing emphasis on the potential significance of the placental microbiome and microbiome–metabolite interactions in immune responses and subsequent pregnancy outcome, especially in relation to preterm birth; however, the existence of a placental microbiome remains open for debate. While studies have shown the presence of commensal bacteria in the placenta, many others have found this to be due to contamination from the environment and/or gene sequencing reagents or methods. It has been suggested that poor pregnancy outcomes result from an inflammatory response originating from the mother, fetus and/or placenta. Dysbiosis of the vaginal, gut or placental microbiome and subsequent alterations to secondary metabolite biosynthesis are important in the development of infection, inflammation and pathogenesis of PTL. Aberrant placental, maternal and FIRs are implicated in premature activation of the pathways involved in labour. However, early determination of placental pathological pathways associated with PTB remains challenging. There is a dearth of functional placental experimental models necessitating further investigation to elucidate the role of the placenta in the molecular pathways involved in gestation-specific initiation of parturition. A comprehensive investigation involving the omics techniques including genomics, transcriptomics, proteomics, metabolomics and lipidomics across different races and ethnic groups is necessary to determine the phenotypes and functions of the placental microbiome and physicochemical signature and the consequent fetomaternal inflammatory (immune) reactions that may lead to maternal antifetal rejection and prematurity. Novel predictive markers of preterm birth may become available based on the understanding of the placenta’s contribution to the timing of delivery.
Take home messages
The existence of a placental microbiome remains open for debate.
The importance of microbiome-metabolite interactions and associated immune responses is rapidly emerging.
Evidence of these interactions and immune reponses contributing to adverse pregnancy outcomes, including preterm birth, is growing.
Acknowledgments
NIHR Global Health Research Group on PReterm bIrth prevention and manageMEnt (PRIME) at The University of Sheffield, UK; University of Cape Town, South Africa; University of Pretoria, South Africa; and The International Centre for Diarrhoeal Disease Research (icddr,b), Bangladesh. https://www.primeglobalhealth.co.uk
References
Footnotes
Handling editor Tony Mazzulli.
Presented at A portion of this work was presented at the Sheffield Global Health Symposium as a short oral presentation in January 2020.
Contributors The review idea was conceptualised by all authors. EA and KMP performed the literature search and produced the initial draft. All authors contributed to the subsequent drafts and critical revision of the manuscript with KMP and EA leading the process. All authors read and approved the final draft before submission for publication.
Funding This review was supported by funding from the National Institute for Health Research (NIHR, 17/63/26).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.