Article Text

Abstract
For a nationwide real-word data study on the application of predictive mutation testing of patients with colorectal cancer (CRC) for anti-epidermal growth factor receptor (EGFR) therapy stratification, pathology data were collected from the Dutch Pathology Registry from October 2017 until June 2019 (N=4060) and linked with the Netherlands Cancer Registry. Mutation testing rates increased from 24% at diagnosis of stage IV disease to 60% after 20–23 months of follow-up (p<0.001). Application of anti-EGFR therapy in KRAS/NRAS wild-type patients was mainly observed from the third treatment line onwards (65% vs 17% in first/second treatment line (p<0.001)). The national average KRAS/NRAS/BRAF mutation rate was 63.9%, being similar for next-generation sequencing (NGS)-based approaches and single gene tests (64.4% vs 61.2%, p=ns). NGS-based approaches detected more additional potential biomarkers, for example, ERBB2 amplifications (p<0.05). Therefore, single gene tests are suitable to stratify patients with mCRC for anti-EGFR therapy, but NGS is superior enabling upfront identification of therapy resistance or facilitate enrolment into clinical trials.
- colorectal neoplasms
- pathology
- molecular
- diagnostic techniques and procedures
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Treatment for patients with metastatic colorectal cancer (mCRC) mainly involves chemotherapy in combination with targeted therapy. Targeted therapy such as anti-epidermal growth factor receptor (EGFR) antibodies (ie, cetuximab and panitumumab) block EGFR activation and hence its downstream signalling cascade.1 However, tumours with mutations in the downstream effectors KRAS or NRAS respond poorly to anti-EGFR therapy.2 Therefore, the KRAS/NRAS mutation status should be evaluated prior to anti-EGFR therapy. Additional potential predictive biomarkers for resistance to anti-EGFR therapy include BRAF p.V600E, PIK3CA mutations and ERBB2 amplifications.3 4 Moreover, BRAF p.V600E mutations are associated with a poor prognosis of patients with mCRC.5 6
Several (inter)national clinical practice guidelines have been developed to provide evidence-based recommendations to assist in the treatment and management of patients with mCRC.7–10 Predictive biomarker analysis has been recommended for a decade now. However, uptake of KRAS mutation analysis to stratify patients for anti-EGFR therapy ranges between 35% and 69%.11–15 We performed a nationwide real-world data study on the application of predictive mutation testing of patients with CRC for anti-EGFR therapy selection and analysed the results of multi-institutional mutation analyses using different analytical methods, performed between October 2017 to June 2019. In addition, mutation testing rates and uptake of anti-EGFR therapy were studied in the last quarter of 2017.
Methods
Databases
Clinical and pathology data were obtained from data linkage between the Netherlands Cancer Registry (NCR) and the nationwide network and registry of histopathology and cytopathology in the Netherlands (PALGA).16 Both databases cover the whole Dutch population (~17 million inhabitants). All newly diagnosed malignancies are registered in the NCR. A standardised structured dataset is collected from patient records consisting of basic patient and disease characteristics, including age, gender, histology, tumor-node-metastasis stage, performance score, the site(s) of metastasis, and type of first-line treatment. All pathology reports from the 46 Dutch pathology departments are digitally archived in PALGA.
Data collection mutation analyses
Diagnostic yield of mutation analysis was studied from pathology reports of patients with CRC that were collected from PALGA using specific queries from 1 October 2017 to 30 June 2019. Manual curation of these reports showed 4060 patients with CRC undergoing predictive mutation analyses in this 21-month study period. Details of the mutation analyses (ie, technique, gene panel, diagnostic yield) were manually extracted from these reports.
Data collection mutation testing rates
To study uptake of molecular testing, all patients that were initially diagnosed with stage IV CRC between 1 October 2017 and 31 December 2017 were selected from the NCR (N=653). A trusted third party (ZorgTTP17) linked these data in September 2019 to PALGA, which was successful for 620/653 (94%) patients.
Data collection uptake anti-EGFR therapy
Uptake of anti-EGFR therapy was examined in patients with CRC that underwent molecular testing between 1 October 2017 and 31 December 2017 (N=547). A trusted third party17 linked these pathology reports from PALGA to the NCR, which was successful for 511/547 (93%) patients. In this linked dataset, mutation-tailored uptake of anti-EGFR therapy was studied. Four hundred eighty-six patients harboured metastatic disease. For 402 of these patients, all lines of treatment were registered, while for 84 patients it was only known whether anti-EGFR therapy was applied or not. Maintenance or partial modification (eg, due to toxicity) of therapy was not considered as new treatment line.
Statistical analysis
Associations between variables were studied using the Fisher’s exact test using IBM SPSS Statistics (V.25). Two-sided p-values are reported. Turnaround time was compared using the Mann-Whitney U test. Funnel plots were generated to study interpathology department variation in diagnostic yield.18 Differences in mutation testing rates over time were studied using the McNemar test.
Results
Multi-institutional molecular characterisation of CRC cases
Mutation analysis results were obtained of 4060 patients with mCRC (figure 1A). Forty-eight per cent of these patients harboured a (likely) pathogenic mutation in KRAS, 5% in NRAS and 12% in BRAF (figure 1B). The KRAS p.G12C mutation was reported in 3.4% of the patients, for which a small molecule inhibitor recently showed encouraging anticancer activity in a phase I trial.19 Mutation analyses were performed by 23 different pathology departments. Limited variation was observed in KRAS/NRAS/BRAF mutation rates among pathology departments (figure 1B,C, online supplemental figure 1A). One department reported a significantly higher frequency (69.4%; p=0.026) and one department a significantly lower frequency (55.3%; p=0.04) than the national average (63.9%). The median turnaround time from initial request of the molecular analysis until authorisation of the pathology report was 7 working days.
Supplemental material

Molecular characterisation of patients with colorectal cancer (CRC). (A) Timeline and flow chart showing the search strategy to obtain all nationwide pathology reports of mutation analysis of patients with CRC performed between 1 October 2017 until 30 June 2019. A large proportion of reports was excluded as no mutation analyses were present (ie, the queries ‘molecular biology’, ‘MSI’ (microsatellite instability) and ‘MLH1’ obtained high amount of reports with immunohistochemistry of mismatch repair proteins). Manual curation of obtained pathology reports showed 4060 patients with CRC undergoing predictive mutation analyses in this 21-month study period. (B) Frequency of reported KRAS, NRAS and BRAF mutations obtained from the pathology reports depicted in panel A. (C) Funnel plot showing the reported diagnostic yield (ie, percentage of KRAS, NRAS and BRAF mutations) per pathology department. Nationwide percentage and 95% CI are shown. (D) Frequency of reported KRAS, NRAS and BRAF mutations by multigene panel next-generation sequencing (NGS) and non-NGS analysis. (E)Mutational landscapes of CRC cases concerning KRAS, NRAS, BRAF, PIK3CA and ERBB2 alterations. Results of multigene panel NGS approaches are shown left and non-NGS approaches right. Each column represents a tumour sample. Each row represents a gene. A coloured bar represents a variant (see legend), a white bar represents no alteration and a grey bar represents not analysed (ie, not present in NGS panel or single gene analysis).
Eighty-one per cent of the mutation analyses were performed with next-generation sequencing (NGS) using targeted gene panels encompassing mainly hotspot regions of 15–75 genes (online supplemental figure 1B,C). All methods covered at least exons 2 and 3 of KRAS and NRAS, and exon 15 of BRAF. Neither in overall reported mutation frequencies of KRAS, NRAS and BRAF nor in the number of failed analyses significant differences between NGS and the remaining techniques were observed (figure 1D). Studying the different mutation types in more detail showed enriched detection of BRAF mutations other than codon 600 by multigene panel NGS (1.7% vs 0.5%, p=0.018; figure 1E). Also (likely) pathogenic PIK3CA mutations (10% vs 2%), (likely) pathogenic ERBB2 mutations (0.6% vs 0%) and ERBB2 amplifications (0.6% vs 0%) were enriched on NGS analyses (p<0.05). These differences were due to more comprehensive testing of NGS-approaches. An overview of reported variants in other genes in the NGS panels is shown in online supplemental figure 2.
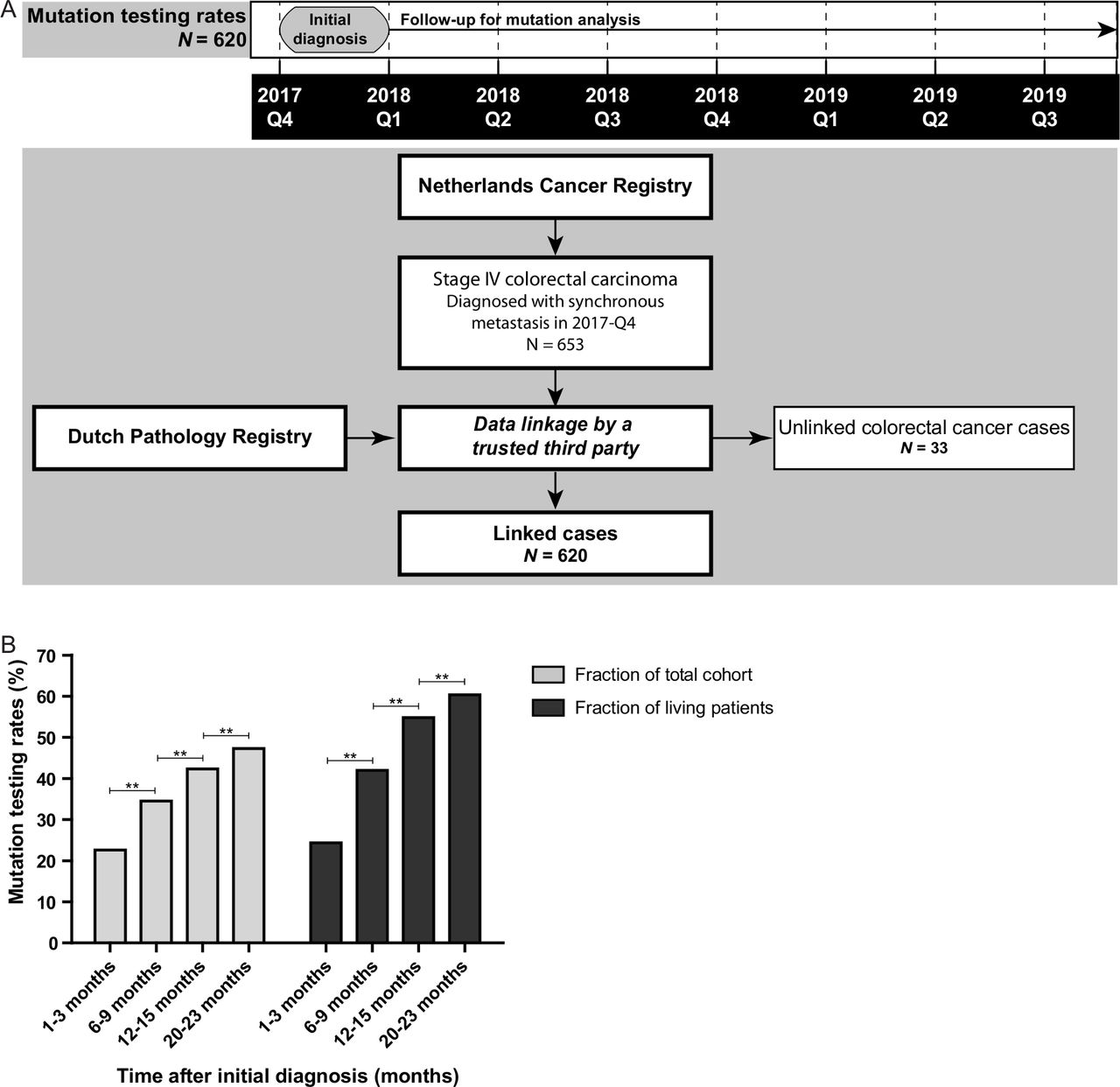
Mutation testing rates of patients with mCRC
Uptake of mutation analysis was studied in patients with mCRC that were diagnosed with stage IV disease in the fourth quarter of 2017 (figure 2A). Mutation testing rates gradually increased over time from diagnosis, that is, from 24% after 1–3 months of follow-up to 60% after 20–23 months of follow-up (p<0.001; figure 2B). Uptake of mutation analysis was enriched in patients diagnosed with mCRC below age 70 years, left-sided tumours, metastases in multiple organs (M1b), metastasis in peritoneum only (M1c), moderately differentiated tumours, and a good performance status (online supplemental table 1 and figure 3).
Supplemental material

Mutation testing rates of patients with metastatic colorectal cancer (CRC). (A) Timeline and flow chart showing the data collection of the pathology reports of all patients with CRC diagnosed with synchronous metastasis between 1 October and 31 December 2017. These patients were obtained from the Netherlands Cancer Registry and linked to the Dutch Pathology Registry (PALGA) by a trusted third party in September 2019. This linked dataset was used to study the mutation testing rates. Unlinked patients with CRC were mainly caused by absence of a pathological diagnosis. (B)The uptake of mutation analysis for patients with stage IV CRC after different periods of follow-up: 1–3 months, 6–9 months, 12–15 months and 20–23 months of follow-up. Data are shown for the whole cohort (N=620) and corrected for patients alive 1 month after initial diagnosis (N=571), 6 months after diagnosis (N=436), 12 months after diagnosis (N=321) and after 21 months (N=227). The McNemar test was applied to study significance. **P <0.001.
Mutation informed targeted therapy choices
Uptake of anti-EGFR therapy was studied for patients with mCRC that underwent mutation analysis in 2017-Q4 (figure 3A). Forty-seven point nine per cent (233/486) of the patients with mCRC were reported to have no mutations in KRAS or NRAS and hence were candidates for anti-EGFR therapy. Thirty-three per cent (76/233) of these patients indeed received anti-EGFR therapy. Uptake was 17% (23/135) for patients without KRAS or NRAS mutations that received one or two lines of treatment compared with 67% (42/63) for patients without KRAS or NRAS mutations that received three or more lines of treatment (p<0.001; figure 3B,C). Anti-EGFR therapy was applied in 4.9% (4/81) mCRC with mutations in KRAS or NRAS (N=3 codon 12; N=1 codon 13), and in 7.4% (6/81) of cases with a BRAF p.V600E mutation.

{kind=link}
{kind=link}
{kind=link}
Uptake of anti-epidermal growth factor receptor (EGFR) therapy in patients with metastatic colorectal cancer (mCRC). (A) Time line and flow chart showing the data collection of both synchronous and metachronous mCRC that underwent mutation analysis between October and December 2017. These patients were linked to the Netherlands Cancer Registry by a trusted third party, enabling therapy registration of these patients. Twenty-five of the patients were excluded for further analysis, as they were diagnosed with stage I–III CRC in 2017 and hence were no candidates for anti-EGFR therapy. Patient records were fully updated including specification of all treatment lines for patients with diagnosis of the primary tumour from 2015 onwards (N=402). For patients with a primary tumour before 2015 (N=84), registration was limited to whether anti-EGFR therapy was applied or not. Unlinked cases were caused by limitations in the linkage procedures. (B) Uptake of anti-EGFR therapy in patients without a KRAS or NRAS mutation merged over all treatment lines (left). In the right panel of the graph uptake for patients that received one or two treatment lines is compared with patients receiving three or more treatment lines. NB: for 35 patients without a KRAS or NRAS mutation it was only registered whether anti-EGFR therapy was applied or not. The Fisher’s exact test was applied to study differences in molecular testing rates. **P<0.001. (C) Overview of the proportions of patients that were treated with anti-EGFR therapy per treatment line. The number of patients present in each line is depicted on top of each bar.
Discussion
Mutation analyses of patients with mCRC generated consistent KRAS/NRAS/BRAF mutation rates independent of the analytical method, showing that both multigene NGS panels and single gene tests can be used to stratify patients with mCRC for anti-EGFR therapy. The current number of predictive biomarkers for mCRC is modest and all molecular targets required for currently registered targeted therapeutics can be adequately assessed by single gene tests combined with immunohistochemical analysis of mismatch repair proteins and NTRK fusions. Nevertheless, multigene panel NGS is a more comprehensive alternative, if used cost-effectively, enabling upfront identification of patients with rare alterations linked to anti-EGFR therapy resistance (eg, ERBB2 amplifications) and rare driver mutations that facilitate enrolment into clinical trials.
Limited variation was observed in KRAS, NRAS and BRAF mutation rates among the 23 pathology departments that performed mutation analyses as only two departments showed aberrant mutation frequencies compared with the national average. However, large variation was observed in the number of predictive biomarker tests performed per laboratory, ranging from less than 1–30 analyses per month, which limited the comparative power.
Unlike international guidelines,7–9 the Dutch guidelines in 2017 recommended to use anti-EGFR therapy only in later treatment lines.10 This recommendation likely explains our observations that molecular testing rates increased over time after diagnosis of stage IV disease and anti-EGFR therapy was mainly applied in later treatment lines. In turn, these observations indicate that a selective, costs-saving approach was used to perform mutation analysis to select patients for anti-EGFR therapy. Nevertheless, mutation testing rates of 60% suggest that anti-EGFR is not considered for all patients with mCRC. These data are in line with previously reported studies, showing an uptake of KRAS testing varying between 35% and 69%.11–15 New guidelines as well as the availability of promising therapy for patients with BRAF p.V600E and KRAS p.G12C mutant mCRC will likely accelerate mutation testing rates.19–22
The use of real-world clinical and pathology data on a nationwide scale shows the strength of linkage of both the NCR and PALGA. However, the use of existing registries also necessitates accepting limited data collection due to predefined collection strategies. In the current study design, we could not confirm that all patients with mCRC receiving anti-EGFR therapy were evaluated for presence of KRAS, NRAS and BRAF mutations before start of the therapy. This requires attention in future research as inferior response rates for patients with KRAS mutations and high costs of anti-EGFR therapy compared with the costs of mutation analysis, clearly necessitate mutation analysis for patients who are candidates for anti-EGFR therapy.23–26
In conclusion, nationwide real-world data show that hotspot mutations in KRAS, NRAS and BRAF for stratification of anti-EGFR therapy in patients with mCRC were consistently detected by both multigene NGS panels and single gene tests, which were selectively applied depending on the treatment line. Combined with immunohistochemical analysis of mismatch repair deficiency proteins and NTRK fusions, these genomic alterations encompass all molecular biomarkers to stratify patients with mCRC for currently registered targeted therapeutics. Multigene panel NGS is a comprehensive alternative for single gene tests if used cost effectively or to identify patients with CRC for clinical trials.
Ethics statements
Patient consent for publication
Ethics approval
The study was conducted in accordance with the Declaration of Helsinki and approved by the Local Ethical Committee of the Radboudumc (CMO 2016–2967). All data were handled according to the General Data Protection Regulation.
Acknowledgments
The authors thank the pathology departments that participate in the PATH project, Riki Willems for the annotation of the pathology data, and the registration team of the Netherlands Comprehensive Cancer Organisation (IKNL) for the collection of data for the Netherlands Cancer Registry.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Runjan Chetty.
Collaborators PATH consortium members: P Drillenburg, E W P Nijhuis: Onze Lieve Vrouwe Gasthuis, Amsterdam; M J van de Vijver, C J van Noesel: Amsterdam UMC, locatie AMC, Amsterdam; E Bloemena, D A M Heideman, T Radonic, E Thunissen: Amsterdam UMC, locatie VUmc, Amsterdam; P M Nederlof, G A Meijer, K Monkhorst: Antoni van Leeuwenhoek, Amsterdam; H Doornewaard: Gelre ziekenhuis, Apeldoorn; M C R F van Dijk, E Ruijter: Rijnstate, Arnhem; K Duthoi: Pathologisch en Cytologisch Laboratorium Amphia, Breda; C Meijers: Reinier de Graaf Arash, Delft; A J C van de Brule, P T G A Nooijen: Jeroen Bosch Ziekenhuis, Den Bosch; F J Bot: Hagaziekenhuis, Den Haag; H M Hazalbag, P Clahsen: Medisch Centrum Haaglanden/ Bronovo, Den Haag; F H van Nederveen, P J Westenend: Laboratorium voor Pathologie, Dordrecht; J W M Jeuken: Stichting PAMM, Eindhoven; E J M Ahsmann, P Meulbroek: Groene Hart Ziekenhuis, Gouda; W Timens, E Schuuring, L C van Kempen: Universitair Medisch Centrum Groningen, Groningen; W Geuken: Martini Ziekenhuis, Groningen; N W J Bulkmans, F E Bellot: Spaarne Gasthuis, Haarlem; R Clarijs: Zuyderland Medisch centrum, Sittard-Geleen; S Riemersma, R van der Geize: Laboratorium Pathologie Oost Nederland, Hengelo; J Meijer, H J van Slooten: PALGA, Houten; R E Kibbelaar, E M J van der Logt: Pathologie Friesland, Leeuwarden; T van Wezel, H Morreau, J V M G Bovee: Leids Universitair Medisch Centrum, Leiden; A Zur Hausen, E J M Speel: Maastricht Universitair Medisch Centrum, Maastricht; P C de Bruin, C J Huijsmans: St. Antonius Ziekenhuis, Nieuwegein; S Dusseljee, R W Willems: Radboud universitair medisch centrum, Nijmegen; C F Prinsen, S Zomer: Canisius-Wilhelmina Ziekenhuis, Nijmegen; F J van Kemenade, W N M Dinjens, W R R Geurts-Giele: Erasmus MC, Rotterdam; H van der Valk, A J J Smits, K J Hoogduin: Pathan BV, Rotterdam; M Kliffen, M A den Bakker: Maasstad ziekenhuis, Rotterdam; J Stavast: Elisabeth ziekenhuis, Tilburg; P J van Diest, W W J de Leng: Universitair Medisch Centrum Utrecht, Utrecht; A P de Bruïne: VieCuri Medisch Centrum, Venlo; J.W J Hinrichs, C Meischl: Symbiant BV, Alkmaar/Hoorn/Den Helder/Zaandam; J Gaal, M Niemantsverdriet: Isala, Zwolle.
Contributors EMPS, KG and MJLL were involved in the study design. GRV and MAGE were involved in the data collection from the Netherlands Cancer Registry. QJMV was involved in the data collection from PALGA. PATH consortium members reported turnaround times and were available for questions. EMPS was involved in the data extraction and annotation from the pathology reports, and performed the data analyses. EMPS, GRV, MAGE, HG, IDN, KG and MJLL interpreted the data and drafted the manuscript. The manuscript was revised and approved by all authors.
Funding This work was supported by the research programme Personalised Medicine of the Netherlands Organisation for Health research and Development (ZonMw, project number 846001001).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.