Article Text

Abstract
Background Paxillin is a modular protein that localises to cell adhesion sites where it facilitates bidirectional communication between the intracellular actin cytoskeleton and the extracellular matrix. These complex and dynamic interactions are essential for cell adhesion, cell migration and cell survival. The authors have previously demonstrated that paxillin is overexpressed in lung cancer tissues and identified somatic paxillin mutations in 9% of lung cancers. A murine in vivo xenograft model of the most common paxillin mutation (A127T) showed increased cell proliferation and invasive tumour growth, establishing an important role for paxillin in the development of lung cancer.
Methods The authors analysed 279 bronchoscopy-aided biopsy specimens from 92 high-risk patients. Adenocarcinoma with bronchioloalveolar features and pure bronchioloalveolar carcinoma (BAC) were analysed with fluorescence in situ hybridisation (FISH) and immunohistochemistry (IHC).
Results Paxillin is overexpressed in premalignant areas of hyperplasia, squamous metaplasia and goblet cell metaplasia, as well as dysplastic lesions and carcinoma in high-risk patients. Concordance between increased paxillin gene copy number and paxillin overexpression was observed in cases of adenocarcinoma eusomic for chromosome 12.
Conclusions Paxillin overexpression occurs during the earliest stages of lung cancer development. FISH and IHC analysis of lung adenocarcinoma suggests that relatively small-scale genomic rearrangements of chromosome 12 are associated with paxillin overexpression in lung adenocarcinoma.
- Lung cancer
- paxillin
- high-risk patient
- premalignant lesion
- preinvasive lesion
- cytogenetics
- adenocarcinoma
- bronchioloalveolar carcinoma
- cancer
- cell adhesion molecules
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Lung cancer
- paxillin
- high-risk patient
- premalignant lesion
- preinvasive lesion
- cytogenetics
- adenocarcinoma
- bronchioloalveolar carcinoma
- cancer
- cell adhesion molecules
Introduction
Lung cancer is the leading cause of cancer death in the USA. Most often, lung cancer is diagnosed when the disease is advanced, resulting in limited therapeutic options and corresponding poor survival times. At the same time, advances in early detection through the use of low-dose spiral CT imaging clearly enhance the ability to detect lung cancer at early stages when therapeutic intervention can potentially extend life.1 2 Autofluorescence bronchoscopy (AFB) is a potential method for identifying high-grade dysplasia and carcinoma in situ within the central airway.3–6 However, AFB has low positive predictive value and low specificity, since biopsy interpretation is confounded by the fact that not all premalignant and preinvasive lesions progress to lung cancer,7 and low-grade dysplasia, inflammation, hyperplasia, metaplasia and granulation tissue all have abnormal AFB patterns.8 Hence, despite improvements in early lung cancer detection, it is not yet evident that early detection by CT or AFB results in mortality reduction. Therefore, discovering clinically important lung cancer biomarkers to identify patients at high risk of developing lung cancer is critical to improving lung cancer diagnosis and treatment.
We recently identified somatic mutations in the gene paxillin9 in lung cancer. The paxillin gene codes for a 68 kDa focal adhesion molecule which is recruited to nascent cell adhesion sites upon activation of αβ integrin heterodimers.10 Paxillin serves as a scaffold for additional structural proteins and regulatory proteins that collectively promote changes in actin dynamics leading to regulation of cell adhesion, migration and survival. These protein–protein interactions are regulated by phosphorylation of paxillin via a large number of kinases providing a mechanism for dynamic control of the assembly and disassembly of large macromolecular complexes.11 Tyrosine phosphorylation of paxillin indirectly activates members of the Rho family of G-proteins leading to the formation of filopodia, lamellipodia and stress fibres, which are all actin-based cellular structures localised at the leading edge of migrating cells.12 Paxillin promotes cell survival via its interaction with Bcl-29 13 14 during development, and paxillin and Bcl-2 colocalise in lung cancer tissue.9 Paxillin is often overexpressed in lung cancer, suggesting that it may contribute to malignancy and serve as a useful biomarker for diagnosis of lung cancer and a cellular target for lung cancer treatment.
In order to further characterise the role of paxillin during lung cancer development and assess its utility as a biomarker, we examined paxillin expression and localisation in bronchial biopsy specimens from patients at high risk of developing lung cancer. We observed early and persistent overexpression of paxillin during lung cancer development in high-risk patients. As paxillin overexpression is observed in both early neoplastic lesions and lung cancer, we used fluorescence in situ hybridisation (FISH) to demonstrate that paxillin gene copy number is often amplified in lung adenocarcinoma and pure bronchioloalveolar carcinoma (BAC). We also show that increased paxillin gene copy number and paxillin protein concentrations correlate with chromosome 12 eusomy.
Methods
Patient and sample selection
The high-risk patients and corresponding biopsy samples examined in this study have been previously described.15 Patients' clinical and pathological information was obtained from a lung bronchoscopy database (Roswell Park Cancer Institute) and a lung cancer patient database (University of Chicago) according to each institutional review board protocol. The patients seen at Roswell Park did not have cancer at the time they were evaluated with bronchoscopy. They had at least two risk factors for cancer: previous history of lung cancer with no evidence of disease for >2 years before the current biopsy; diagnosis of chronic obstructive pulmonary disease; asbestos-related lung disease16; >20 pack-year cigarette smoking history. These comprehensive databases include clinical information and risk factor data such as age, gender, smoking status, medical history, occupational exposure, pathological diagnosis, tumour stage (TNM (tumour/nodes/metastasis)) and tumour grade.
Histological examination
H&E-stained slides of bronchial biopsy samples from 105 patients were reviewed by two pathologist (ACM and MT) and categorised according to World Health Organization (WHO) classifications.17 Of these, 92 contained sufficient respiratory mucosa to be evaluated. ‘Premalignant’ changes include hyperplasia, squamous metaplasia and goblet cell metaplasia. ‘Preinvasive’ epithelial lesions include atypical adenomatous hyperplasia (AAH), dysplasia and carcinoma in situ.
Immunohistochemistry
Tissue microarray (TMA) paraffin sections were deparaffinised in xylene, rehydrated through graded ethanol solutions to distilled water, and then washed in Tris-buffered saline. Antigen retrieval was carried out by heating sections in ETDA buffer (pH=9) for 15 min in a microwave oven. Endogenous peroxidase activity was quenched by incubation in 3% H2O2 in methanol for 5 min. Non-specific binding sites were blocked using Protein Block (Dako, Carpinteria, California, USA) for 20 min. Tissue sections were incubated for 1 h at room temperature with the mouse monoclonal paxillin antibody (clone 5H11, dilution 1:200; Neomarkers, Freemont, California, USA) followed by 30 min incubation with goat anti-mouse IgG conjugated to a horseradish peroxidase-labelled polymer (Envision+ System; DakoCytomation, Carpinteria, California, USA). Slides were developed for 5 min with 3,3′-diaminobenzidine chromogen and counterstained with haematoxylin. Negative controls were performed by replacing the primary antibody step with non-immune mouse immunoglobulins. Immunostaining intensity in the cytoplasm was scored as follows: 0=no staining; 1=weak staining; 2=moderate staining; and 3=strong staining. In this study, immunohistochemistry (IHC) scores of 2 and 3 were both interpreted as overexpression. For comparison between means of two groups, Student t test or χ2 test was used.
KRAS and EGFR mutation analysis
Sections 4 μm thick were cut from formalin-fixed, paraffin-embedded tissue samples and placed on to membrane-coated laser capture microscopy slides (LCM; Leica, Wetzlar, Germany.). Slides were deparaffinised in xylene, rehydrated through gradient ethanol, stained with H&E, and dehydrated in ethanol. Target lesions were identified and isolated by LCM. Tissue fragments were digested overnight in proteinase K (10 mg/ml), DNA was purified using Qiagen MinElute kits, and resuspended in 10 mM Tris/HCl, pH 8.0.
Point mutations in KRAS codons 12 and 13 were identified using a Light Cycler 2.0 as described by Nikiforova et al.18 In brief, purified DNA was used to generate a PCR amplicon using primers (K-rasF1: 5′-AAGGCCTGCTGAAAATGACTG; K-rasR1: 5′-GGTCCTGCACCAGTAATATGCA) that flank the KRAS codon 12/13 mutational hotspot. Using a pair of fluorescently labelled anchor and sensor oligonucleotides (KRASanc1: 5′-CGTCCACAAAATGATTCTGAATTAGCTGTATCGTCAAGGCAGT-fluorescein; KRASsnsr1: 5′-LC Red640-TGCCTACGCCACCAGCTCCAA-phosphate), amplification and mutation detection was accomplished via fluorescence resonance energy transfer. The sensor oligonucleotide spans the mutation site allowing the detection of a codon 12 or codon 13 mutation based on the distinct melting temperatures (Tm) of wild-type KRAS and mutant KRAS. DNA from a cell line carrying a mutation in KRAS codon 13 served as a positive control.
EGFR mutation was detected by sequencing genomic DNA purified as described for KRAS mutation detection. Exons 19 and 21 were amplified by PCR using the following exon flanking primer pairs: 19F, 5′-CCTGAGGTTCAGAGCCATGGAC, and 19R, 5′-CAGCATGTGGCACCATCTCACAATTGC; and 21F, 5′-CTTCCCATGATGATCTGTCCCTCACAGC, and 21R, 5′-GGAGAGCATCCTCCCCTGCATGTGT. Amplicon sizes were checked by agarose electrophoresis, which permitted detection of large deletions. The amplicons were purified by ethanol precipitation, sequenced using BigDye terminator sequencing chemistry run on an ABI 3100 capillary analyser. The resulting DNA sequences were analysed with Mutation Surveyor V3.23 software.
Tissue microarray
The study group consisted of 51 wedge resections and lobectomies performed at the University of Chicago Medical Center from 1990 to 2008. These cases were divided into three groups: (1) invasive adenocarcinoma with BAC pattern, (2) ‘pure BAC’ without invasive adenocarcinoma, and (3) adenocarcinoma without BAC. Eleven of the pure BAC cases were non-mucinous type (92%). There were lymph node metastases in three cases of invasive adenocarcinoma. A morphologically representative area of interest within each paraffin donor block was identified by two pathologists (AHN and MT) under the microscope using an H&E-stained section on a glass slide as a guide. From each case a minimum of five tissue cylinders with a diameter of 1 mm were arrayed into a recipient block using an automated tissue microarrayer ATA-27 (Beecher Instruments, Sun Prairie, Wisconsin, USA). In the case of adenocarcinoma with BAC pattern, the cores were obtained as follows: two cores from invasive tumour, two from BAC component, and one from adjacent normal lung. In the case of pure BAC or invasive adenocarcinoma without BAC, five tissue cylinders included three cores from the tumour and two cores from adjacent normal lung. A total of 270 tissue cores were arrayed on to the two recipient TMA blocks, which were then cut into 4 μm thick serial sections and subjected to IHC analysis.
Fluorescent in situ hybridisation and analysis
Two-colour FISH analysis was performed using the BAC probe RP11-144B2, which localises to 12q24.23 and contains the full-length paxillin gene, directly labelled with Spectrum Orange using a Nick Translation Kit (Abbott Molecular, Des Plaines, Ilinois, USA), together with CEP 12 directly labelled with Spectrum Green (Abbott Molecular), which hybridises to the alpha-12 centromeric region (12p11.1–12q11.1).19 Slides were pretreated using the Paraffin Pretreatment Kit (Abbott Molecular). At least 18 interphase nuclei were analysed for each patient. Only single, clearly defined nuclei were scored; overlapping nuclei or clusters of nuclei were not analysed. Split fluorescence signals with similar intensity and diameter seen in the same focal plane adjacent to each other were counted as one (eg, see figure 3F). For most cases, small round diploid nuclei likely to be infiltrating lymphocytes were used as an internal control.
Paxillin and chromosome 12 gene copy number were determined by dividing the total number of fluorescent signals corresponding to paxillin (RP11–144B2) or alpha-12 (CEP 12), respectively, by the total number of nuclei analysed for each patient. Since paxillin gene copy number was heterogeneous within individual tumour samples, equivocal cases of paxillin gene copy number alteration were not included in our analysis if we failed to observe individual nuclei with more than five copies of paxillin or if nuclei with more than two copies of paxillin represent <30% of the total nuclei scored. FISH was validated by scoring 437 and 417 normal nuclei for paxillin and alpha-12, respectively. In this report, the averaged number of fluorescent signals in normal nuclei was 2.04 for paxillin and 1.85 for alpha-12, and these averages were used for all subsequent analysis of malignant nuclei. Unpaired Student t test was used to determine statistical significance for each case: the copy number of paxillin and alpha-12 from malignant nuclei measured from an individual case was compared with the average copy number from all normal nuclei (ie, 2.04 and 1.85 for paxillin and alpha-12, respectively); this was done for every case analysed. When possible, the average gene copy number of paxillin and alpha-12 from malignant nuclei measured from an individual case was compared with the average copy number of paxillin and alpha-12 from normal nuclei measured from the same corresponding case. In all instances, both methods were in complete statistical agreement. For paxillin, unpaired Student t test between normal and malignant nuclei with p<0.01 was interpreted as a gene copy number alteration; for alpha-12, unpaired Student t test between normal and malignant nuclei with p<0.01 was interpreted as chromosome 12 polysomy. Paxillin amplification was defined as a paxillin gene copy number increase and chromosome 12 eusomy. ‘Polysomy’ was defined as a copy number increase in both paxillin and chromosome 12. Loss of heterozygosity was defined as a decrease in paxillin gene copy number and chromosome 12 eusomy.
Results
Histological changes and paxillin expression in patients at high risk of developing lung cancer
From the initial cohort of 105 patients at high risk of developing lung cancer (table 1), biopsy material from 92 patients was suitable for histological and paxillin analysis. This resulted in a total of 279 individual biopsy samples for analysis (table 2) with between one and six (mean number of three) biopsy specimens analysed for each patient. No histological abnormalities were observed on any of the individual biopsy samples of 10 patients; the remaining patients had one or more biopsy specimens containing a portion of histologically normal appearing respiratory mucosa. Hence, most patients (89%) had one or more foci of histologically abnormal mucosa.
Patient characteristics (n=105)
Distribution of histological findings by patient (n=105) and biopsy (n=279)
A total of 173 histological normal biopsy specimens were analysed for paxillin expression by IHC. In 74 of 92 patients (80%), no paxillin expression was observed in histologically normal surface epithelium (see figure 2A of Jagadeeswaran et al9), whereas weak, moderate and strong paxillin expression was observed in 13 (14%), four (5%) and one (1%) patients, respectively (data not shown). This showed that paxillin was either not detectable or was weakly expressed in histologically normal appearing respiratory mucosa in ∼95% of the high-risk patients, which is similar to our previous findings.9
An increase in paxillin expression in biopsy samples containing areas of abnormal histology was observed (figure 1). Whereas 11% of all histologically normal appearing biopsy samples showed moderate to strong concentrations of paxillin, moderate to strong paxillin expression was observed in 78%, 81% and 93% of the hyperplastic, squamous metaplastic and goblet cell metaplastic lesions, respectively. Paxillin expression appeared strongest in the basal layer (arrows, figure 1). Four of the 92 patients (4%) had areas of dysplasia (figure 2A,B) and three (3%) had lung carcinoma (figure 2C,D). All of these patient samples showed elevated levels of paxillin expression, and expression appeared strongest along the basal layer at the peripheral margins of the preinvasive lesions (black arrows, figure 2D). Together, these findings indicate that, relative to adjacent normal respiratory mucosa, paxillin expression was upregulated in non-neoplastic precursor lesions before malignant changes were microscopically evident and can remain elevated during the formation of preinvasive epithelial lesions.

Paxillin is overexpressed in premalignant epithelial lesions in high-risk patients. Representative images of transbronchial biopsy samples stained for paxillin. Paxillin staining localises to the cytoplasm and is enriched in the basal layers of the respiratory mucosa (arrows). The bottom row of pie graphs describes quantitative paxillin staining intensity in biopsy samples with hyperplasia, squamous metaplasia or goblet cell metaplasia morphology.
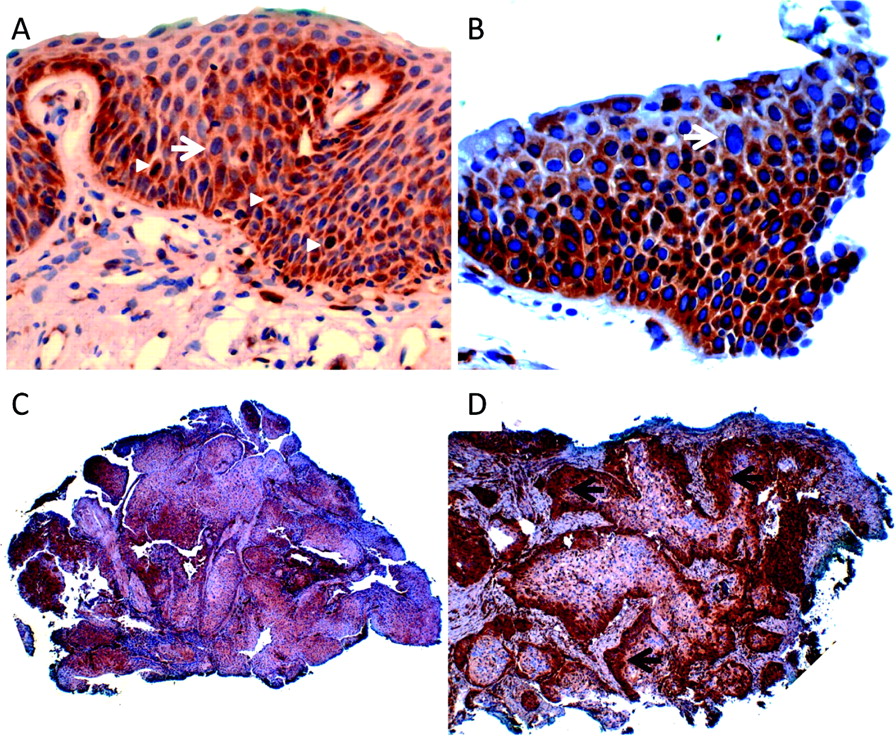
Preinvasive epithelial lesions from high-risk patients overexpress paxillin. Bronchoscopic biopsy samples showing dysplasia (A,B) and carcinoma (C,D). Note the large, irregular nuclei in the upper portion of the respiratory (white arrow) and mitotic figure (arrowheads) mucosa in (A) and (B). Paxillin staining is strongest along the basal and parabasal layers (arrows in D).
Dysplasia and carcinoma were associated with a long history of smoking (table 3). On the other hand, hyperplasia, squamous metaplasia and goblet cell metaplasia were associated with a corresponding increase in mean smoking pack-year history, indicating that hyperplastic changes occur earliest in smokers, whereas goblet cell metaplasia occurs in the setting of a longer smoking history. The highest levels of paxillin staining were observed in patients with the longest smoking history. Stronger paxillin staining was visible in 69% of biopsy samples from current smokers (22/32) compared with 49% of biopsy samples from former smokers (24/49) (p=0.0395, χ2 test).
Pack-years of smoking by histology and paxillin expression
Gene copy number of paxillin correlates with histological subtype and KRAS mutation
A pilot series of 11 human non-small cell lung cancer (NSCLC) samples representing various histological subtypes were examined by FISH to determine if paxillin (12q24.23) gene copy number alteration correlated with a specific histological subtype of lung cancer (table 4). Paxillin gene copy number was increased in two of four adenocarcinoma samples, whereas paxillin gene copy number changes in other histological subtypes were not observed. EGFR and KRAS mutation status were examined on enriched tumour foci isolated using laser capture microscopy. A KRAS mutation in codon 12/13 was detected in one of the two adenocarcinomas cases with increased paxillin gene copy number, indicating that KRAS muation20 21 and altered paxillin gene copy number are not mutually exclusive. No mutations in exon 19 or exon 21 of EGFR in any samples were detected.
Correlation of paxillin copy number (FISH) with EGFR (exon 19/21) and KRAS (codons12/13) mutation
Paxillin locus is amplified in adenocarcinoma including pure BAC subtype
On the basis of the relatively high frequency with which paxillin gene copy number alterations were identified in lung adenocarcinoma in the initial small set of samples, together with our previous finding of paxillin amplification in human lung cancer cell lines,9 FISH was used to examine paxillin copy number in 39 cases of adenocarcinoma comprising 27 cases of adenocarcinoma with a BAC component and 12 cases of pure BAC (figure 3). Compared with normal nuclei with two copies of paxillin (figure 3A), 13/27 cases of adenocarcinoma (48%) (table 5) and 5/12 cases of pure BAC (42%) (table 6) showed altered paxillin gene copy number. With the exception of two cases of pure BAC (see below), all tumours showed increased paxillin gene copy number. Three to six copies of paxillin were typically observed (figure 3B–F); however, occasionally individual nuclei with up to 16 fluorescent signals corresponding to the paxillin locus were observed. Two of three cases with lymph node metastases showed increased paxillin gene copy number similar to the corresponding primary tumour. Occasionally, individual tumour specimens showed heterogeneous alterations in paxillin gene copy number such that regions with increased paxillin copy number were adjacent to regions with normal copy number.

Paxillin gene copy number is increased in lung adenocarcinoma and bronchioloalveolar carcinoma (BAC). Representative DAPI (4',6-diamidino-2-phenylindole)-stained nuclei showing FISH probes for paxillin (red) and alpha-12 (green). Non-malignant nuclei exhibit two fluorescent signals for both paxillin and alpha-12 (A). Primary lung adenocarcinoma measuring <5 mm (B), 5–10 mm (C), >10 mm (D), pure BAC (E) and metastatic adenocarcinoma (F).
Correlation of increased paxillin gene copy number with chromosome 12 and paxillin expression in lung adenocarcinoma with BAC component
Correlation of paxillin copy number variation with chromosome 12 and paxillin expression in pure bronchioloalveolar carcinoma lung cancer
As chromosomal instability is common in NSCLC,22 23 chromosome 12 centromeric region was co-analysed to determine if increased paxillin gene copy number was a non-specific consequence of chromosome 12 polysomy (see the Methods section). For adenocarcinoma, 8/13 cases (62%) with increased paxillin gene copy number showed chromosome 12 polysomy, and 5/13 cases (38%) showed chromosome 12 eusomy (table 5) (p=0.217, χ2 test). Of the five cases of pure BAC with altered paxillin gene copy number, three showed increased paxillin gene copy number and two demonstrated loss of heterozygosity for paxillin (table 6). Of the cases of adenocarcinoma with a BAC component, none showed loss of heterozygosity for paxillin. We did not observe any tumour specimen with multiple copies of chromosome 12 centromeric regions and two or fewer copies of paxillin.
Paxillin protein concentrations were determined by IHC on 36/39 tumour specimens that were analysed by FISH (figure 4). Alterations in paxillin gene copy number were seen in 17/36 cases, and five of these cases also showed paxillin overexpression (table 5). Further analysis showed that 4/5 cases with paxillin amplification (ie, chromosome 12 eusomy) showed paxillin overexpression (ie, cases 2, 6, 8 and 9 in table 5), whereas 1/7 cases with chromosome 12 polysomy showed paxillin overexpression (ie, case 11 in table 5) (p=0.046, Fisher exact test). This finding suggests that paxillin overexpression in lung adenocarcinoma is more likely to occur with relatively small alterations in chromosome 12 that include the paxillin locus, compared with genomic rearrangements involving the entire chromosome 12.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Paxillin is overexpressed in adenocarcinoma and bronchioloalveolar carcinoma (BAC). Representative H&E and paxillin-stained tissue microarray tissue cores from cases of adenocarcinoma with BAC component and pure BAC are shown along with the distribution of paxillin staining intensity.
Discussion
This is the first analysis of paxillin expression during the earliest stages of lung cancer development. By analysing paxillin expression in 279 biopsy specimens from 92 patients at high risk of developing lung cancer, we demonstrate that paxillin is overexpressed in the majority of premalignant lesions such as hyperplasia, squamous metaplasia and goblet cell metaplasia. We also observed that paxillin is overexpressed in preinvasive epithelial lesions. We found that paxillin gene copy number is often increased in the bronchioloalveolar subtype of lung adenocarcinoma and pure BAC. We further identified a subset of human lung adenocarcinoma—characterised by relatively small-scale rearrangements of chromosome 12 affecting the paxillin locus—where paxillin overexpression correlated with increased paxillin gene copy number.
On the basis of our previous work,9 we hypothesised that paxillin becomes overexpressed in premalignant lesions when histological evidence of neoplasia is first evident. Current models suggest that lung cancer develops through a progressive continuum of histological changes in normal respiratory mucosa towards advancing grades: hyperplasia, squamous and/or goblet cell metaplasia, dysplasia and AAH/carcinoma in situ.7 24 These changes are in turn driven by an underlying accumulation of molecular alterations that ultimately lead to microinvasive NSCLC. Although these lesions are associated with the development of NSCLC in smokers and other high-risk individuals and are often identified in lung resections, they do not invariably progress, and instead often regress after smoking cessation.25 This finding underscores the variability in biological behaviour of these lesions and emphasises the need to identify biomarkers with diagnostic, prognostic and therapeutic utility. We observe that paxillin is widely overexpressed in most premalignant lesions of varying histological morphology. Since most of these lesions will not go on to develop into lung cancer, our findings indicate that paxillin overexpression is not a specific biomarker for identifying premalignant lesions in high-risk patients.
Instead paxillin overexpression probably reflects a role related to proliferation and survival associated with altered or damaged respiratory epithelium secondary to smoking-related injury. This is supported by the relative increase in paxillin expression in the basal and parabasal layers and along the leading edges of invasive lesions. Reserve progenitor cells are believed to reside in the basal layer, and smoking elicits changes in gene and protein expression in the epithelium of smokers associated with injury response and proliferation.26 Protein concentrations of Ki-67, a proliferation marker expressed in dividing cells but not resting cells, measured in bronchial biopsy samples of current and former smokers showed a similar expression pattern to paxillin27 with enhanced expression along the basal and parabasal layers. Interestingly, Ki-67 was more highly expressed in current smokers than former smokers. We also measured lower paxillin expression in former smokers compared with current smokers. However, the significance of this observation is uncertain, as we were unable to compare paxillin expression in former smokers with biopsy material obtained when they actively smoked. Increased paxillin expression is also observed in glomerular immune injury, suggesting upregulation of integrin signalling in response to accumulation of extracellular matrix proteins and scarring.28 Together, these findings support a role for paxillin during epithelial response to injury in patients with a history of exposure to cigarette smoke.
Paxillin is a scaffold protein composed of five N-terminal LD motifs, four C-terminal LIM domains, and several SH3 domains which allow paxillin to form a large number of protein–protein interactions. The LIM3 domain targets paxillin to focal adhesion sites, thereby regulating its subcellular localisation. Consequently, paxillin is able to integrate multiple signalling pathways within the cell by providing docking sites for common signalling molecules and positioning them adjacent to activated receptors such as integrin heterodimers and growth factor receptors at the cell membrane.11 It would therefore appear beneficial for a cell to express paxillin during wound healing in response to injury29 as well as during malignant transformation.30–32 For example, the terminal LD4 domain of paxillin binds the BH4 domain of Bcl-2, and this interaction promotes survival and morphogenesis during kidney development.13 14 We have previously shown that paxillin colocalises with Bcl-2 in NSCLC.9 This raises the intriguing hypothesis that paxillin overexpression in premalignant lesions promotes survival via Bcl-2-mediated signalling. This interaction in turn can facilitate tissue repair, as well as tumour development. Nakanishi et al33 demonstrated increasing levels of Bcl-2 expression in low-grade to high-grade AAH lesions and non-mucinous BAC. Although we did not directly examine paxillin and Bcl-2 colocalisation in the biopsy specimens, it is tempting to speculate that this interaction may be an early event in lung cancer development, and future studies can test this prediction.
Consistent with our previous work9 and the analysis presented here, little to no paxillin expression is observed in histologically normal respiratory epithelium. This is in marked contrast with liver, where paxillin expression shows no statistically significant difference in tumour and adjacent non-tumour liver tissue,31 indicating that baseline levels of paxillin expression in normal cells are tissue dependent. We also observed a significant correlation between paxillin overexpression and increased paxillin gene copy number in a subset of lung adenocarcinoma with a diploid complement of chromosome 12. This finding suggests alternative mechanisms for paxillin overexpression depending on the tumour genotype. In cases of polysomy for chromosome 12, the lack of correlation between paxillin gene copy number and paxillin overexpression suggests that additional mutations in other key genes develop during tumorigenesis, at which point paxillin overexpression becomes dispensable. KRAS is located on chromosome 12 and is a known driver mutation in lung cancer, raising the possibility that paxillin overexpression is no longer necessary when tumours become polysomic for chromosome 12. Interestingly, we only observed paxillin loss of heterozygosity in pure BAC tumours, which also failed to overexpress paxillin. In summary, this is the first description of paxillin overexpression in the earliest morphological changes leading up to the development of lung cancer. We also identify a subset of lung adenocarcinoma in which changes in paxillin gene copy number correlate with paxillin overexpression, and future studies will address the clinical significance of this observation.
Take-home messages
Paxillin expression is upregulated in non-neoplastic precursor lesions before malignant changes are microscopically evident, suggesting that paxillin plays a role related to proliferation and survival associated with altered or damaged respiratory epithelium.
48% of adenocarcinoma and 42% of pure bronchioloalveolar carcinoma showed altered paxillin gene copy number, and cases with lymph node metastases showed increased paxillin gene copy number similar to the corresponding primary tumour.
Concordance between increased paxillin gene copy number and paxillin overexpression was observed in a subset of lung adenocarcinoma with a diploid complement of chromosome 12.
Acknowledgments
ACM is grateful to Dr Elizabeth McNally, Dr Eric Svensson, Dr Kaul and Dr Nowak for support.
References
Footnotes
Funding Supported in part by NIH/National Cancer Institute (5R01CA125541-03, 3R01CA125541-03S109, 5R01CA129501-02, 3R01CA129501-02S109, 5P01HL058064-140009), V-Foundation (Guy Geleerd Memorial Foundation), Kate McMullen Foundation, Respiratory Health Association of Chicago, and Mesothelioma Applied Research Foundation (Jeffrey P Hayes Memorial Grant).
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Roswell Park Cancer Institute IRB and University of Chicago IRB.
Provenance and peer review Not commissioned; externally peer reviewed.