Article Text

Abstract
Polycythaemia vera (PV), essential thrombocythemia (ET) and idiopathic myelofibrosis (MF), are the most common myeloproliferative neoplasms (MPN) in patients without the BCR-ABL1 gene rearrangement. They are caused by clonal expansion of haematopoietic stem cells and share, as a diagnostic criterion, the identification of JAK2V617F mutation. Classically, when other clinical criteria are present, a JAK2V617F negative case requires the analysis of Exon12_JAK2 for the diagnosis of PV, and of MPL515K/L mutations for the diagnosis of ET and MF. Here, we evaluated 78 samples from Brazilian patients suspected to have MPN, without stratification for PV, ET or MF. We found that 28 (35.9%) are JAK2V617F carriers; from the 50 remaining samples, one (2%) showed an Exon12_JAK2 mutation, and another (2%) was positive for MPLW515L mutation. In summary, the investigation of JAK2V617F, Exon12_JAK2 and MPLW515K/L was relevant for the diagnosis of 38.4% of patients suspected to have BCR-ABL1-negative MPN, suggesting that molecular genetic tests are useful for a quick and unequivocal diagnosis of MPN.
- Cancer Genetics
- Haemato-Oncology
- Haematopathology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Polycythaemia vera (PV), essential thrombocythemia (ET), idiopatic myelofibrosis (MF) and BCR-ABL1 chronic myeloid leukaemia (CML) are distinctive haematopoietic diseases from the group of the myeloproliferative neoplasms (MPN). Clinical and biological features set CML apart from PV, ET and MF, and these three entities have some important characteristics in common. They are all caused by the clonal expansion of a multipotent haematopoietic progenitor cell, maintain a relatively preserved haematopoietic differentiation, develop some degree of bone marrow fibrosis, and can evolve into each other or transform into more aggressive haematological cancers.1 In PV, the oncogenic process targets a multipotent precursor committed to the erythroid lineage, causing an expansion of the red cell mass in the peripheral blood and a loss of response to the physiologic regulation of erythropoiesis. In MF, the target is a pluripotent haematopoietic precursor whose transformation leads to an intense stromal proliferation in the bone marrow, and a variety of peripheral blood manifestations.2 In ET, the oncogenic process results in megakaryocytic hyperplasia in the bone marrow and increased platelet counts in the peripheral blood.1 Despite such distinctive phenotypic features, PV, MF and ET can share some important findings at the molecular level, as the identification of a shared clonal marker. Molecular studies have identified a point mutation in the JAK2 gene on PV, MF and ET. The JAK2V617F mutation leads to the constitutive activation of the JAK2 kinase that can be found in 95% of patients with PV, and in 50–60% of patients with MF and ET.3 Due to such high frequencies, the JAK2V617F mutation has become one of WHO's criteria for the diagnosis of PV, MF and ET. Although the JAK2V617F mutation has been also described with low frequencies in different groups of haematological malignancies,4 its identification in cells from the peripheral blood in the appropriate clinical context is virtually diagnostic of a BCR-ABL1-negative MPN. On the other hand, a negative test does not definitely exclude PV, MF and ET in the presence of other clinical and haematologic features of MPN. In this scenario, further molecular investigations are suggested, as the search for mutations in the Exon 12 of the JAK2 gene (Exon12_JAK2), or in the position 515 of the MPL gene (MPLW515K/L). Exon12_JAK2 mutations have been found in approximately 2–3% of PV patients; those mutations are clustered between codons 533 and 547, where they disrupt the pseudo-kinase domain, promoting its constitutive activation.4 MPL gene is predicted to be a haematopoietic cytokine receptor acting through JAK2 intracellular transduction,5 and the MPLW515L mutation results in a constitutive activation of JAK-STAT signalling. Another mutation in this position—MPLW515K—was later identified, but its precise effects on signalling are not yet explained. The 515 tryptophan (W515) is the key amino acid located in a unique amphipathic domain that prevents spontaneous activation of MPL,6 and either MPLW515K/L mutations meet the WHO diagnostic criterion for ET or MF.
Although major diagnostic criteria for MPN are based on clinical features, blood cell counts, hormone levels and bone marrow morphology, these data are not always available or sufficient to conclude a diagnosis. In this regard, novel tools that are fast, convenient and widely available could be helpful in this clinical setting. Thus, the present work was designed to evaluate the frequency of JAK2V617F, Exon12_JAK2 and MPLW515K/L mutations in Brazilian patients clinically suspected to have MPN.
Material and methods
We analysed 78 samples from patients suspected to have MPN, all of which were sent to our clinical laboratory to be tested for the JAK2V617F mutation over a 2-month period. DNA was extracted automatically through a QIACUBE system (QIAGEN) and evaluated using TaqMan-based real-time PCR method. Only the wild type JAK2 samples (JAK2V617F negative) were analysed for Exon12_JAK2 mutations (Sanger sequencing) and for MPLW515K/L mutations (TaqMan-based real-time PCR assay). Primers used are shown in table 1.
Primers and probes used for screening
Real-time PCR reactions were run in a ABI 7900HT (Life Technologies) for JAK2V617F and in a Rotor-Gene 6000 (QIAGEN) for MPLW515K/L. Exon12_JAK2 was sequenced in a 3130 Genetic Analyzer (Applied Biosystems). Reactions parameters, cycling conditions and reagents volumes were used as universal conditions defined by the manufactures.
The study protocol was approved by the Internal Review Board, and all samples were anonymised from patient identifiers for the purposes of this study. To compare our data against previous studies, we assumed that, in the studies in which the stratification of MPNs were presented, the sum of PV, MF and ET patients represent the total of MPN samples, and this number was used as a denominator to identify the frequency of JAK2V617F-positive cases in the cohort.
Results
From the 78 analysed samples, 50 (64%) were negative and 28 (35.9%) positive for JAK2V617F mutation. JAK2V617F mutation frequencies from the present study and those selected from the literature are presented in table 2.
Detailed frequency of JAK2V617F mutation in different cohorts, stratified by PV, MF and ET, or not (*)
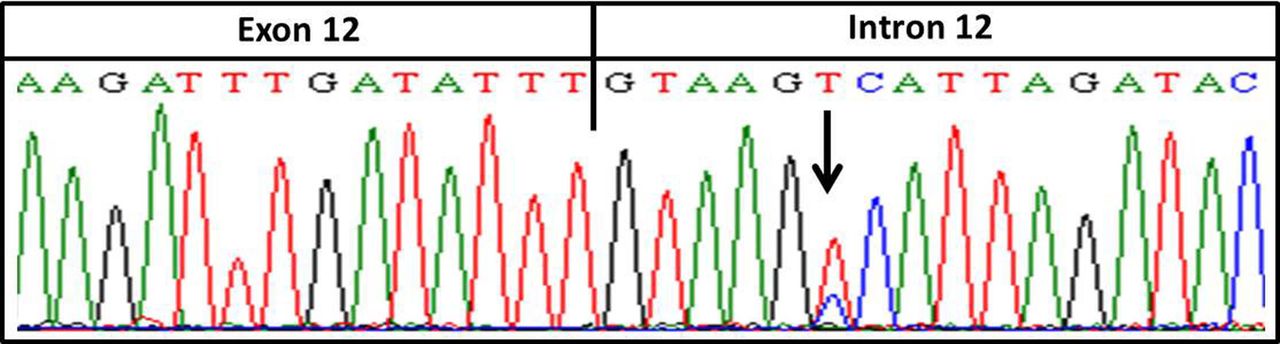
The 50 JAK2V617F negative samples were evaluated for both Exon12_JAK2 and MPLW515K/L mutations. A single patient was identified with an Exon12_JAK2 mutation (c.1194+6T>C) (figure 1). This alteration, deposited as rs182123615, is only six nucleotides apart from the Exon12_JAK2 hotspot area, and is located in a splicing site region.
{kind=link}
Mutation identified on Exon12_JAK2 flanking region. The mutation is six nucleotides apart from the hotspot mutation area.
Analysis of MPL515K/L mutations also identified a single carrying patient where the tryptophan is exchanged by a leucine (W515L). The mutation MPLW515K was not identified in our series.
Taken together, JAK2V617F, Exon12_JAK2 and MPLW515L mutations were present in 38.4% of the cases suspected to have MPN from our cohort. Although JAK2V617F is the most frequent mutation, it is important to note that the other mutations correspond to 4% of JAK2V617F-negative samples (table 3).
Mutation frequency on MPN samples
Discussion
JAK2V617F frequencies in previous studies are higher than in our series (table 2), even when Brazilian patients were evaluated.7–12 A possible reason for this could be the stratification of PV, MF or ET patients as performed by others. Prestratification for PV, ET or MF generally increases the pretest probability of a positive result, given that in this context molecular genetic tests are frequently ordered on a confirmatory basis. In non-stratified cases, on the other hand, these tests are ordered in a broader clinical context, with a lower pretest probability of a MPN diagnosis. This situation is quite common in the clinical laboratory setting, where molecular genetic tests are frequently ordered for patients with sustained high blood counts in a ‘rule out MPN’ approach. The probability of a positive test in this setting is, therefore, much lower than that for prestratified cases, as we have observed in our series. Kiladjian et al13 used a similar approach in their study, without prestratification for PV, ET or MF, and observed similar frequencies. However, no MPLW515K/L or Exon12_JAK2 mutations were identified in their study, despite the higher number of patients evaluated. In our series, those mutations corresponded to 2% of JAK2V617F-negative cases, or 1.28% of all cases suspected to have MPN clinically.
The analysis of Exon12_JAK2 identified a single mutation carrier. Although no clinical data was related to this mutation in the COSMIC, HGMD, LOVD, HGVS, OMIM and ClinVar databases, it is present in a splice site region, probably leading to constitutive activation of the JAK2 protein. In the 1000 genome database, it is present in 0.1% of the overall population, being identified solely in Americans, but not in African, Asian and European individuals (0.002). Although further clinical data would be invaluable to better characterise this finding, the retrospective and anonymised design of our study has prevented this approach. In summary, our study suggests that molecular genetic tests for JAK2V617F, Exon12_JAK2 and MPL515K/L mutations are relevant for the investigation of patients suspected to have BCR-ABL1-negative MPN.
Take-home messages
-
Molecular tests emerge as a convenient tool to quickly confirm the diagnosis in patients suspected to have BCR-ABL1-negative myeloproliferative neoplasms (MPN).
-
JAK2V617F, Exon12_JAK2 and MPLW515K/L molecular analysis are useful for the investigation of cases suspected to have BCR-ABL1-negative MPN even without polycythaemia vera, essential thrombocythemia or myelofibrosis prestratification.
-
In our series, a test panel evaluating JAK2V617F, Exon12_JAK2 and MPLW515K/L was relevant to confirm MPN diagnosis in 38.4% of BCR-ABL1 negative MPN cases.
Acknowledgments
We would like to thank José de Sá for his great contribution on data retrieval; Karla de Oliveira Pelegrino for helping with MPL and Exon12_JAK2 cloning; and Danyella Silva Pereira for helping with DNA extractions.
References
Footnotes
MTdS and MM-N contributed equally
-
Contributors MTdS, MM-N, KM designed the study, performed the experiments and drafted the manuscript. MdLLFC and EGR contributed as clinical haematologists. EGR revised the manuscript. All authors read and approved the final version.
-
Funding This work was supported by FINEP (02.12.0223.00).
-
Competing interests None.
-
Ethics approval Fleury Internal Review Board.
-
Provenance and peer review Not commissioned; externally peer reviewed.