Article Text

Abstract
Aims Some types of monoclonal gammopathies are typified by a very limited availability of aberrant cells. Modern research use high throughput technologies and an integrated approach for detailed characterisation of abnormal cells. This strategy requires relatively high amounts of starting material which cannot be obtained from every diagnosis without causing inconvenience to the patient. The aim of this methodological paper is to reflect our long experience with laboratory work and describe the best protocols for sample collection, sorting and further preprocessing in terms of the available number of cells and intended downstream application in monoclonal gammopathies research. Potential pitfalls are also discussed.
Methods Comparison and optimisation of freezing and sorting protocols for plasma cells in monoclonal gammopathies, followed by testing of various nucleic acid isolation and amplification techniques to establish a guideline for sample processing in haemato-oncology research.
Results We show the average numbers of aberrant cells that can be obtained from various monoclonal gammopathies (monoclonal gammopathy of undetermined significance/light chain amyloidosis/multiple myeloma (MM)/MM circulating plasma cells/ minimal residual disease MM—10 123/22 846/305 501/68 641/4000 aberrant plasma cells of 48/30/10/16/37×106 bone marrow mononuclear cells) and the expected yield of nucleic acids provided from multiple isolation kits (DNA/RNA yield from 1 to 200×103 cells was 2.14–427/0.12–123 ng).
Conclusions Tested kits for parallel isolation deliver outputs comparable with kits specialised for just one type of molecule. We also present our positive experience with the whole genome amplification method, which can serve as a very powerful tool to gain complex information from a very small cell population.
- HAEMATO-ONCOLOGY
- METHODOLOGY
- MYELOMA
- DNA
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Monoclonal gammopathies (MGs) reflect conditions in which abnormal amounts of immunoglobulins are produced by an abnormal clone that developed from a single pro-B germ cell.
The past decades have seen great strides in the diagnosis and treatment of MGs, and it is now being debated that even multiple myeloma (MM) might represent a potentially curable cancer.1 Clinical research is going deeper and deeper into the smallest populations of cancer cells which form an abnormal clone. Characterisation of specific target antigens and/or mutations of the founder cancer clone in monoclonal gammopathy of undetermined significance (MGUS), light chain amyloidosis (ALA) and residual clone in the minimal residual disease (MRD) stage of MM could shed light on malignant transformation and the chemoresistant progress of the disease.2 ,3
High throughput technologies bring new opportunities to translational genomics research. Unfortunately, these techniques require relatively high amounts of starting material which cannot be obtained from every diagnosis without inconvenience to the patient. It becomes especially difficult where several samples for simultaneous methods are required, analysing different levels of cell organisation (ie, genomic, epigenomic, transcriptional, protein, etc).
Thus isolation of sufficient amounts of nucleic acids of optimal quality for further use represents an extremely challenging issue when conducting research in the field of MGs or other disorders with low amounts of target cells. Overcoming this methodological limitation is a crucial step for successful research outcome.
Here we discuss initial processing of biological material, such as cell cryoconservation, separation algorithms, nucleic acids isolation and other procedures, suitable for samples with very small amounts of cells. We also present several algorithms that help to overcome the limitations and find an optimal strategy. Hence the aim of this paper is to present our solutions following long term laboratory experience researching MGs, comprising preprocessing of patient samples and isolation of nucleic acids of good quality and quantity for subsequent high throughput analyses from minor target cell populations.
Methods and results
Biobanking and sorting of target population
Retrospective analyses in various study designs require long term sample collection and storage. We propose three principle strategies for sample biobanking with respect to long term storage of material at different levels of sample processing.
Cryoconservation of end point material (eg, DNA/RNA)
Cryoconservation of middle point material (eg, aberrant plasma cells (aPC))
Cryoconservation of start point material (eg, mononuclear cells (MNCs))
Advantages and disadvantages of each strategy will be discussed below in detail. We focus on the ‘start point’ material cryoconservation strategy, which means freezing the patient's MNCs at the initial step of sample preprocessing. This strategy faces the wide spectrum of subsequent methodological pitfalls but offers greater flexibility in the design of a retrospective study.
Several cryoconservation protocols were compared to define the optimal balance between sample quality, yield, processing time and financial costs. Our laboratory has chosen the following workflow as the most suitable based on our extensive experience.
Sample collection and cryoconservation
Generally, peripheral blood and bone marrow (BM) samples were obtained from patients during routine diagnostic procedures. All studies were approved by the institutional ethics review boards and all individuals provided written informed consent. Buffy coats from healthy donors were kindly provided for this study by the Blood Centre, University Hospital Ostrava.
Initially, MNCs were isolated by density gradient centrifugation. MNCs (aliquots with 10×106 cells) were frozen in 1 mL of cold fetal bovine serum (FBS) containing 10% dimethyl sulfoxide (DMSO) at −80°C in a freezing container, which allowed a rate of cooling of approximately −1°C/min. For long term storage, samples were subsequently transferred into liquid nitrogen at −196°C.
On analysis, samples were thawed in a water bath at 37°C and immediately centrifuged in 10 mL RPMI 1640 medium enriched with 10% FBS. The pellet was re-suspended in staining buffer and samples were prepared for fluorescence activated cell sorting (FACS).
Another popular cryoconservation protocol with different medium compositions (70% RPMI 1640, 20% FBS, 10% DMSO) used in some other studies4 showed comparable results (data not shown). Interestingly, we observed no significant variability in the viability of samples frozen for periods from 3 months to 3 years when the protocols were performed correctly.
Cell sorting
BM aPCs were sorted on a FACS Aria III (Becton, Dickinson and Company). A combination of markers was used to distinguish between normal and aPCs in more than 90% of cases of MGs: CD45-PB, CD38-FITC, CD19-PC7 and CD56-PE (Dako, Beckman Coulter, Cytognos).5 ,6
To exclude dead cells from samples, BD Horizon Fixable Viability Stain 700 was used (figure 1). This fluorescent dye binds to amines which are differentially accessible in live versus dead cells and this leads to differences in the intensity of the fluorescent signal. Target amines are exposed on the surface of live cells, while they are available on the surface and inside of dead cells that have been permeabilised naturally, during the cell death process. Afterwards, live cells can be fixed and permeabilised for subsequent intracellular staining (eg, for kappa, lambda markers) while maintaining a stable viability stain (BD Bioscience, Technical Data Sheet).

Fluorescence activated cell sorting of aberrant plasma cells in light chain amyloidosis. Mononuclear cells were stained using CD45-PB, CD38-FITC, CD19-PC7 and CD56-PE. Live cells were gated using BD Horizon Fixable Viability Stain 700 (gate P1). Doublets discrimination was done using FSC-A and FSC-H (gate P2). Total plasma cells were differentiated from the mononuclear cell gate (gate P3) using CD45+/− and CD38high (gate PC), and aberrant plasma cells were identified using CD19− and CD56− (dots).
The median number of viable MNCs was 53.7% (range 27.9–72.2%; see table 1) from all MNCs undergoing cell sorting (after doublets and debris discrimination). The same population of viable cells can be expressed as median 9.4% (range 1.9–50.5%) from all MNCs before cryoconservation. The differences are caused by loss of cells during sample cryoconservation, re-washing, and debris and doublets discrimination processes. Comparison of median yields of several sample populations in different diagnoses is provided in table 2.
Milestones in sample preprocessing with examples of common values
Infiltration of aberrant plasma cells in different diagnoses
Nevertheless, following nucleic acid isolation algorithms allow us to efficiently utilise samples with very low infiltration of target cell populations for further high throughput downstream applications in research.
Competitive nucleic acids isolation
To find the best algorithm for isolation of nucleic acids from a very limited target cell amount, DNA and/or RNA extraction was performed using a range of commercially available isolation kits based on selective binding of nucleic acids to silica membranes or magnetic beads (table 3). We did not aim to test traditional precipitation protocols as they are less reproducible between samples and laboratories.
Commercially available nucleic acids isolation kits used in our research
RNA isolation kits comparison
RNA isolation kits were compared with each other and with kits for parallel isolation. Comparison of kits was performed on buffy coat samples with defined numbers of cells in the following aliquots: 1, 10, 20, 50, 100, 150 and 200×103 cells sorted by FACS. Aliquots were defined based on previous experience with availability of target cell population in MGs. Samples were tested in duplicate. The average yield from each aliquot is shown in figure 2. RNA concentrations and integrity numbers (RINs) were evaluated using RNA 6000 Pico kit on Bioanalyzer2100 (Agilent Technologies). The concentration can be also measured with RNA HS assay kit on Qubit 2.0 fluorometer (Thermo Fisher Scientific) but this measurement does not allow evaluation of RIN number and we did not use it in our comparisons.

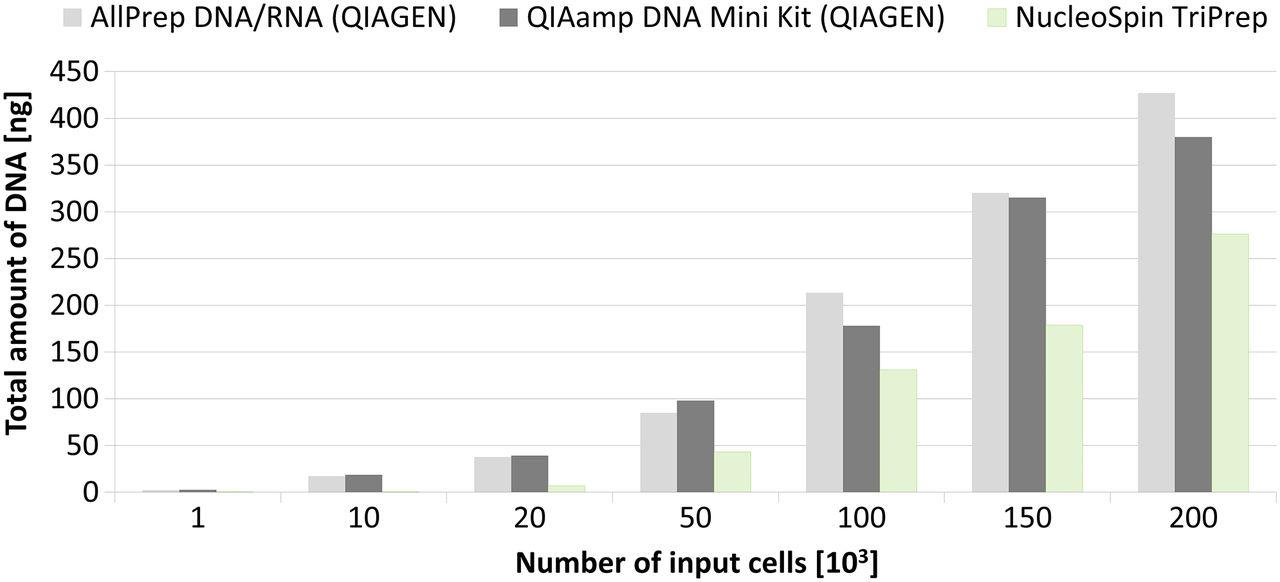
RNA isolation. Mean values of RNA yields after isolation using different kits were obtained from duplicates of aliquots of 1, 10, 20, 50, 100, 150 and 200×103 cells sorted by fluorescence activated cell sorting using forward scatter and side scatter. Both magnetic kits (MagMAX Total RNA and μMACS mRNA) were not sufficient to isolate RNA from a small amount of input material.
Of all of the kits, the NucleoSpin TriPrep (Macherey-Nagel) showed the highest yield but the concentration per microlitre of elution buffer was very low in comparison with AllPrepDNA/RNA Micro Kit (Qiagen) and RNeasy Micro Kit (Qiagen). The main reason was an almost three times higher volume of elution buffer: 40 µL for NucleoSpin TriPrep (Macherey-Nagel) versus 14 µL for AllPrep DNA/RNA Micro Kit and RNeasy Micro Kit (Qiagen). Both magnetic kits (MagMAX Total RNA Isolation Kit and μMACS mRNA Isolation Kit) were not sufficient for successful isolation of RNA from small amounts of input material (figure 2).
Considering that most common downstream applications for RNA, gene expression profiling, requires only a small amount of relatively highly concentrated RNA (eg, 20 ng in 5 µL; RIN>8) and that the concentration of RNA in diluted samples cannot be easily increased due to its sensitivity to further manipulation (decreased integrity) and losses after precipitation, both Qiagen kits were evaluated as more appropriate (table 4).
Comparison of RNA isolation kits
Thus even though total yields of RNA provided by AllPrep DNA/RNA Micro Kit and RNeasy Micro Kit (Qiagen) were more or less comparable, the AllPrep DNA/RNA Micro Kit had an advantage of parallel isolation of both DNA and RNA from one sample and could better exploit a sample with a low number of cells.
Despite the DNA amount being almost constant for all human cell types, transcribed RNA is highly dependent on cell type and its transcriptional activity.
In the case of MGs, the cells of interests are aPC, which highly express paraprotein. To better estimate the yield of RNA from a given number of PCs, we validated the results of the best isolation kit, from our comparison, on PCs from the MM cell line RPMI 8226 (figure 3), and on patient's BM aspirates obtained during routine diagnostic procedures (number of input cells given in table 2, RNA yields not shown). Accordingly, the yields from RPMI 8226 were considered when the diagram in figure 5 was created.

RNA isolation from the multiple myeloma cell line RPMI 8226 using the AllPrep DNA/RNA Micro Kit. Whiskers show SD of the mean values from samples performed in triplicate.
DNA isolation kits comparison
For comparison of DNA isolation efficiency, we used the same design as for RNA. We compared two types of kits, the kit for exclusive isolation of DNA and kits for parallel isolation. DNA concentration was measured using DNA HS Assay on Qubit 2.0 fluorometer (Thermo Fisher Scientific).
Based on the number of input cells and total amount of isolated DNA, two kits (AllPrep DNA/RNA Micro Kit and QIAamp DNA Mini Kit) showed comparable results (figure 4). The AllPrep DNA/RNA Micro Kit was chosen as the best because of its advantage of parallel isolation of two nucleic acids.

DNA isolation. Comparison of two parallel DNA/RNA kits and one DNA isolation kit. Mean values were obtained from duplicates of aliquots of 1, 10, 20, 50, 100, 150 and 200×103 cells sorted by fluorescence activated cell sorting using forward scatter and side scatter.
However, given the fact that one human cell contains approximately 6.6 pg of DNA, our best results achieved only 30% of the theoretical yield. The potential losses were possibly caused by manipulation during DNA isolation as well as by cell cryoconservation and the cell sorting procedure.
Suitability of isolated nucleic acids in downstream applications
Different molecular biology methods require different amounts and quality of input DNA/RNA. Most common applications in current cancer research are PCR, array comparative genomic hybridisation (CGH) and next generation sequencing for DNA, as well as qRT-PCR, gene expression profiling or RNA-seq for RNA, respectively. Based on the known number of aberrant cells along with DNA/RNA yield from different haematological malignancies (ALA, MGUS, MRD), we propose possible applications they can be used for. The workflow is schematically provided in figure 5.
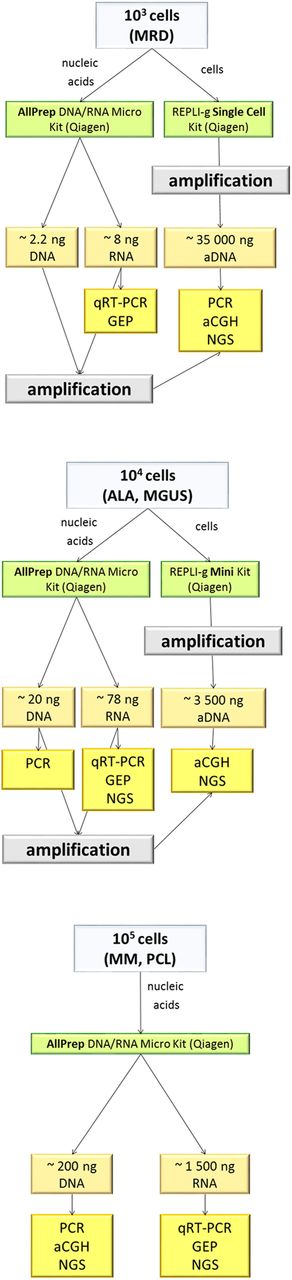
Diagram of preprocessing of samples with small amounts of input material. For a typical number of aberrant cells in various monoclonal gammopathies (MGs), we suggest nucleic acid isolation strategies and expected yield and suitability for downstream applications. For the isolation of nucleic acids with ≤105 cells, the most efficient isolation kit was the AllPrep DNA/RNA Micro Kit. For less starting material or for improved DNA yield for next generation sequencing (NGS), amplification by REPLI-g Single Cell of REPLI-g Mini Kit is recommended. aDNA, amplified genomic DNA; ALA, light chain amyloidosis; CGH, comparative genomic hybridisation; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; MRD, minimal residual disease; PCL, plasma cell leukaemia.
There is a wide range of available protocols for genomic applications, each using different input amounts of DNA. We decided to relate DNA yields to requirements imposed by SureSelect Human All exons v5 kit and SurePrint G3 CGH+SNP Array (4×180 K) (Agilent Technologies, USA) as they are widely used in cancer research. Hence the results from various studies can be easily compared or merged. The minimal threshold for these applications (next generation sequencing, array CGH) is 200 ng, for which we need at least 100 000 cells in the target population. Unfortunately, for ALA, MGUS and MRD, such amounts are scarcely ever reached. For this reason, use of whole genome amplification is needed (see below).
Nucleic acid amplification
Modern high throughput techniques (genome wide/transcriptome wide) require relatively high amounts of starting material. Unfortunately, performance of even the best isolation protocols is limited by the low number of target cells. Thus for some high throughput methods, the required input amount of nucleic acids is so high that the pre-amplification step cannot be avoided. For example, next generation sequencing of aPCs in MM MRD positive patients requires processing of as low as 1×102 cells and whole genome amplification is essential. Whole genomic DNA amplification and whole transcriptome amplification have been implemented in cancer research, especially for analysis at the level of single cells.7 ,8 There are multiple protocols employing different principles available on the market.9
From the set of the most widely used methods (ie, Multiple Annealing and Looping-Based Amplification Cycles (MALBAC), Degenerate Oligonucleotide PCR (DOP-PCR), Primer Extension Preamplification (PEP) and Multiple Displacement Amplification (MDA)) we selected a protocol based on MDA technology because it uses polymerase with the lowest error rate (Phi29 polymerase).9 The Qiagen company developed several modifications of the protocol that differ in the expected reaction yield. It has been previously shown that the REPLI-g Midi kit can be used for amplification of isolated DNA for array CGH,10 allowing amplification from 10 ng DNA to 40 µg per 50 µL without causing a significant bias. We also tested the smaller and cheaper version of this kit (REPLI-g Mini kit) which declares yields of up to 10 µg per 50 µL reaction from 10 ng of input DNA. In our hands, the performance of the latter kit was worse than declared: input DNA ranging from 2.5 to 30 ng was amplified into 1–4 µg (input DNA was obtained using standard AllPrep DNA/RNA Micro Kit (Qiagen) kit).
We wanted to reach an even better amplification rate, and thus we tested the REPLI-g Single Cell Kit, which amplifies DNA directly from cells as well as from isolated genomic DNA. As the high amplification rate (from 2 cells up to 40 µg of DNA) might introduce some errors, we verified DNA amplification specificity and efficiency on 100, 500 and 1000 cells by PCR and quantitative PCR on LightCycler 480 (Roche) on two genes (APOA, TTR) and compared the ratios of the obtained values. Amplification of bulk DNA of a known concentration was used as a control (figure 6). The numbers of cells for this simulation were selected based on the known infiltration of aPCs in MM MRD, the diagnosis with the lowest number of available aberrant cells.
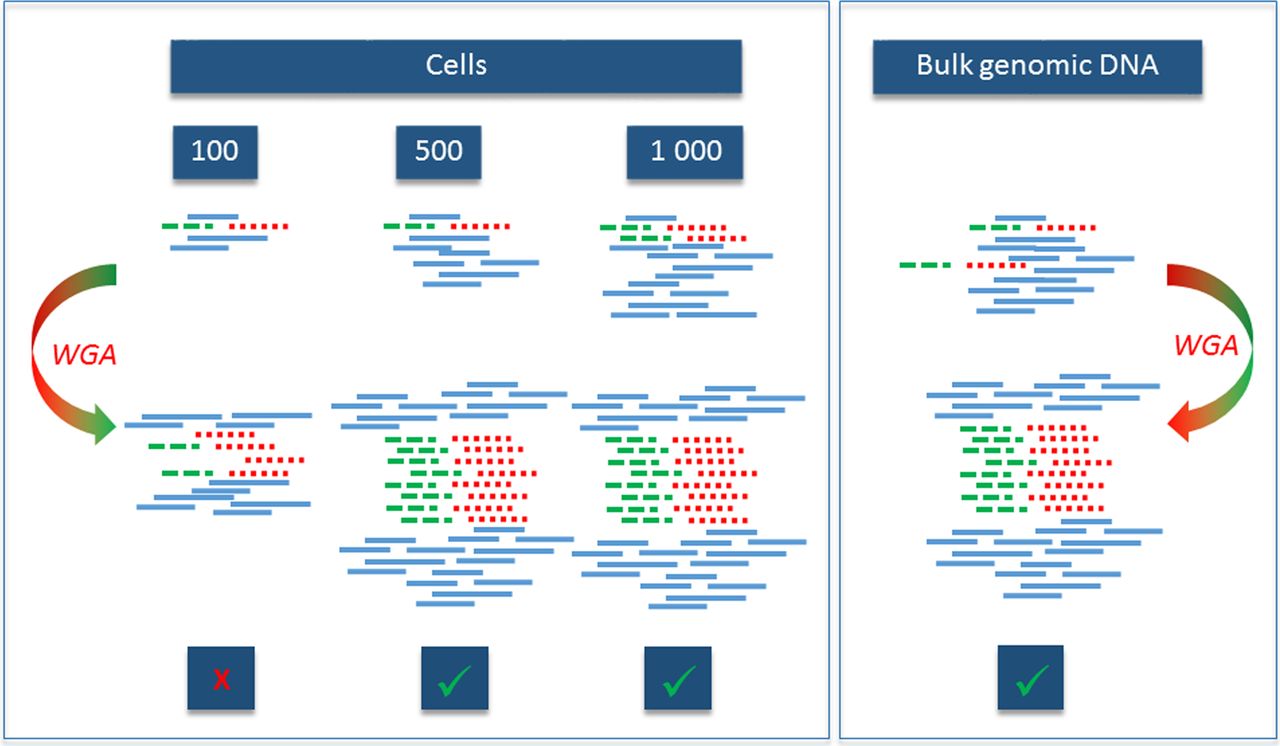
Scheme of amplification efficiency test using the REPLI-g Single Cell Kit. Amplification of two genes was proportional when more than 500 cells were used as a starting point. Broken and dotted lines represent tested genes present in initial and amplified samples among other DNA (full lines).
The results confirmed that the amplification is proportional when more than 500 cells are amplified. Sanger sequencing revealed no mutations in the tested region of the transthyretin (TTR; 18q12.1) gene (500 bp). Thus the threshold for efficient amplification in our conditions was set as 500 cells. The same limit is then applied for routine cell sorting of MRD cells. Amplification was performed according to the manufacturer's recommendations with minor modifications. The reaction volume was decreased to half. The yield of amplified DNA from 5 to 20×102 cells ranged between 13.25 and 19.6 μg, which is 66–98% of expected yield (20 µg per half volume reaction).
Discussion
The design of multicentre studies using high throughput ‘omics’ is confronted with protocol standardisation to avoid batch effects. ‘Omics’ informally refers to a field of study in biology ending in -omics, such as genomics, transcriptomics, proteomics or metabolomics. ‘Omics’ aims at the collective characterisation and quantification of pools of biological molecules that translate into the cell structure, function and metabolism.
Unlike disorders in which the malignant clone tends to dominate the BM at diagnosis (eg, acute leukaemia), in MGs target clones generally represent a very small subset of BM cells.11–13 Thus correct sample preprocessing is essential for all downstream applications in clinical studies as well as in basic research to gain complex information from a small amount of cells.
We have summarised our experience with MG sample processing to create laboratory guidelines. Study of rare MGs meets several methodological limitations.
Essential sample collection and biobanking
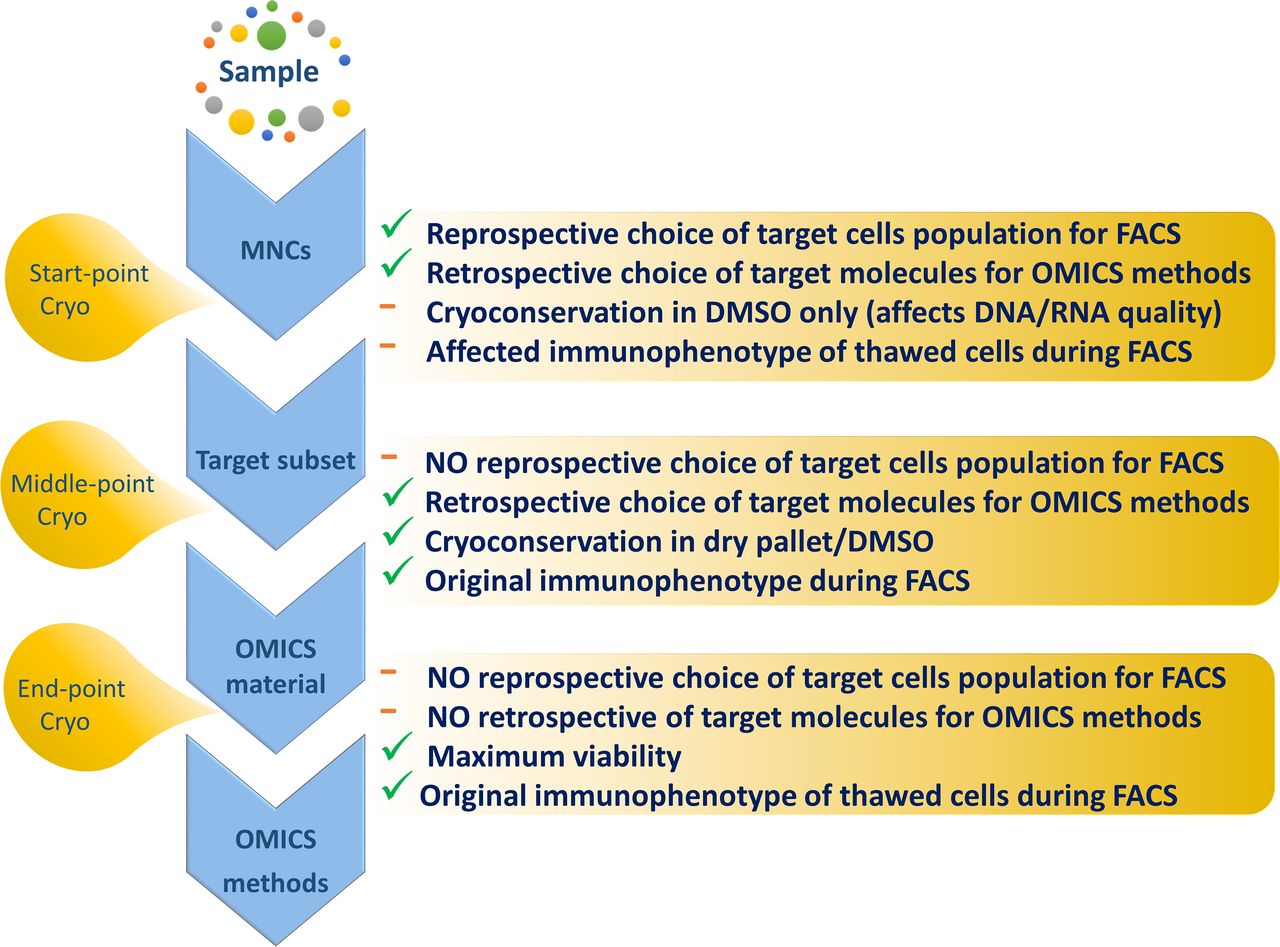
Long term sample collection and biobanking is an essential step for most long term follow-up studies constrained by a retrospective design. As mentioned above, there are three main strategies: cryoconservation of start point, middle point or end point material, respectively.
Undoubtedly, each has its own advantages and disadvantages (figure 7). Cryoconservation of end point material guarantees maximal viability of cells during sample preprocessing and can be suggested as optimal for examination of rare cell subpopulations (eg, MRD study).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of advantages and disadvantages of the three main cell cryoconservation strategies. DMSO, dimethyl sulfoxide; FACS, fluorescence activated cell sorting; MNCs, mononuclear cells.
In addition, the advantage of the last two strategies is an unaffected immunophenotype of target cells after cell sorting. As an illustration, loss of CD138 expression on the PC surface after previous cell cryoconservation was described.14 It is important to note that MNC freezing for further FACS sorting is performed in DMSO solution, which can affect the quality of the final nucleic acids. Thus processing of fresh sample always leads to better quality and higher amounts of final target molecules. However, sometimes such an approach is not feasible for retrospective studies or because of unfavourable time management. Thus despite all the limitations and methodological difficulties, we suppose that in most MGs studies the ‘start point’ strategy (MNC cryoconservation) is preferable because of the flexibility in the design of retrospective studies. This biobanking strategy allows a panel of markers for target population cell sorting and subsequent ‘omics’ screening to be chosen based on long term disease monitoring.
In general, the ‘start point’ approach focuses on indepth targeted description of malignant cell clones in specific cohorts of patients (eg, searching for predictive markers of diseases, potential drug targets and other specific research).15
Target cell sorting with high purity to avoid contamination for subsequent sensitive research techniques
In MGs research, very limited tumour material with a very low infiltration of aberrant cells in BM aspiration is available compared with solid tumours. Under such conditions, manipulation with frozen samples for further sorting and nucleic acids isolation becomes a challenge.
The previously used immunomagnetic separation technique (magnetic activated cell sorting (MACS)) for PC enrichment was limited by material losses and a higher risk of contamination16 but it can still find its place in PC separation protocols as a quick method for pre-enrichment in highly cellular samples with low input infiltration before final cell sorting.17 The threshold for MACS separation is 5% infiltration of PCs—below this value it is not possible to reach high PC purity in sorted cell fractions.16
FACS is a suitable method for separation of specific cell types, such as clonal PCs with infiltration <5% because it allows separation according to light scatter characteristics and more than one fluorescently labelled antibody.17 Moreover, FACS can separate subpopulations with low abundance (under 0.1%) of the initial infiltration.18
Post-sorting sample processing to reach high quality material with minimal losses for downstream application
Nowadays, research objectives require sample analyses on several levels (ie, genomic, transcriptomic, proteomic, etc) simultaneously. Modern molecular research has revealed that important insights can be made through understanding interactions between them.19 Obtaining comprehensive results from high throughput molecular techniques has two methodological limitations: (i) it is crucial to provide good quality and high amounts of nucleic acids or other target molecules and (ii) it is necessary to isolate DNA/RNA simultaneously from the same sample and then perform paired analyses. Evaluation of amount and quality of nucleic acids can be performed in multiple ways, especially for RNA, which is very prone to degradation by Rnases and requires several control steps to secure optimal quality for high throughput techniques as microarrays.20 Some studies have claimed lower RIN values (4–6.6) are sufficient21 while another preferred RIN above 8.22 Our experience with gene expression microarrays and qRT-PCR showed that RIN should be ≥8.
Concentration of nucleic acids is more precisely and more sensitively measured using fluorometry (eg, Qubit) than using absorbance in UV spectrum (eg, Nanodrop). However, a fluorometer does not usually allow evaluation of the purity of the sample. On the other hand, following the manufacturer’s protocols in commercial kits guarantees isolation of nucleic acids of high quality. The problem with reaching minimal input amounts of nucleic acids can be overcome by use of whole genome amplification. Although such an approach can introduce different types of biases, we did not experience any serious negative impact on results in downstream applications. Moreover, emerging single cell techniques might help to overcome all of these limitations in the future but it may always remain economically and technically too demanding for MGs research as the aberrant population is of high genetic heterogeneity and would require many biological replicates in single cell settings.
This work has examined the pitfalls of research related to MGs; some have been successfully overcome while others persist. Establishment, validation and standardisation of thorough laboratory guideline is necessary before initiation of a research project to prevent loss of valuable samples, proper assessment of minor aberrant subclone and isolation of DNA/RNA with sufficient quantity and quality for modern high throughput methods.
This methodological paper has described, based on our knowledge and long experience, the best protocols for sample collecting, sorting and further preprocessing according to the available amount of cells and intended downstream application in MGs research with detailed description of the potential pitfalls.
Take home messages
High throughput techniques require high amounts of starting material. To obtain sufficient amounts of biological material can cause inconvenience to patients with some haematological malignancies.
Correct and unified sample preprocessing guidelines are essential in clinical and basic research to acquire complex information from a limited amount of biological material.
The paper presents several algorithms for sample processing that help overcome methodological limitations and find optimal research strategies.
Acknowledgments
We thank Dr Vyacheslav Yurchenko (Life Science Research Centre Ostrava) for assistance with the qPCR settings. We are also grateful to the team at the University Hospital Brno (Dr Pour), including the Biobank (Dr Almáši) and flow cytometry laboratory (Dr Bezděková) for help to increase the cohorts by providing bone marrow samples from patients with monoclonal gammopathies.
References
Supplementary materials
Abstract in Czech
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Abstract in Czech - Online abstract
Footnotes
TŠ and KG contributed equally to this work.
Correction notice This article has been corrected since it was published Online First. Additional information was added to the acknowledgements section.
Handling editor Mary Frances McMullin
Contributors TS, KG, ZKu, JF and PV performed the experiments. JF, FK and TJ performed the flow cytometry analyses. EK, TS and KG wrote the manuscript. ZKo, FK and RH designed the study and provided the clinical background. TS and KG contributed equally to this study.
Funding This study was supported by the Institutional Development Plan of the University of Ostrava (IRP201550) and the Ministry of Education, Youth and Sports (specific university research of the Faculty of Medicine, University of Ostrava) SGS03/LF/2015–2016 and SGS10/LF/2016–2017, University Hospital Ostrava DRO-FNOs/2016/21, DRO-FNOs/2014/17P and the Ministry of Health of the Czech Republic (15-29667A and 17-30089A).
Competing interests None declared.
Ethics approval The study was approved by the University Hospital Ostrava (66/2016).
Provenance and peer review Not commissioned; externally peer reviewed.