Article Text

Abstract
Aims Amyloidosis is caused by deposition of abnormal protein fibrils, leading to damage of organ function. Hereditary amyloidosis represents a monogenic disease caused by germline mutations in 11 amyloidogenic precursor protein genes. One of the important but non-specific symptoms of amyloidosis is hypertrophic cardiomyopathy. Diagnostics of hereditary amyloidosis is complicated and the real cause can remain overlooked. We aimed to design hereditary amyloidosis gene panel and to introduce new next-generation sequencing (NGS) approach to investigate hereditary amyloidosis in a cohort of patients with hypertrophic cardiomyopathy of unknown significance.
Methods Design of target enrichment DNA library preparation using Haloplex Custom Kit containing 11 amyloidogenic genes was followed by MiSeq Illumina sequencing and bioinformatics identification of germline variants using tool VarScan in a cohort of 40 patients.
Results We present design of NGS panel for 11 genes (TTR, FGA, APOA1, APOA2, LYZ, GSN, CST3, PRNP, APP, B2M, ITM2B) connected to various forms of amyloidosis. We detected one mutation, which is responsible for hereditary amyloidosis. Some other single nucleotide variants are so far undescribed or rare variants or represent common polymorphisms in European population.
Conclusions We report one positive case of hereditary amyloidosis in a cohort of patients with hypertrophic cardiomyopathy of unknown significance and set up first panel for NGS in hereditary amyloidosis. This work may facilitate successful implementation of the NGS method by other researchers or clinicians and may improve the diagnostic process after validation.
- amyloid
- haematology
- DNA
- molecular genetics
- peripheral blood
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Amyloidosis is caused by extracellular deposition of abnormal protein fibrils, resulting in destruction of tissue architecture and damage of organ function.1 Amyloidosis can manifest in different types. In total, 27 different human proteins were described to be connected with amyloidogenic potential in vivo.2 The most common classification reflects the localisation of the amyloid (systemic vs localised) or the disease origin (acquired vs hereditary amyloidosis).3 In systemic amyloidosis, more than one organ or organ systems are affected, while in localised amyloidosis, only one organ or organ system is damaged. Different types of amyloidosis arise from different predisposing conditions: (1) plasma cell dyscrasia might develop into immunoglobulin light chain amyloidosis (AL); (2) long-lasting inflammation can cause reactive amyloidosis (AA); or (3) germinal mutations in certain genes can lead to hereditary amyloidosis (HA).4 5 However, the origin of some other amyloidosis types caused, for example, by prion protein PRNP or integral membrane protein 2B ITM2B has not been elucidated, despite the ongoing research in this area.
Combination of several factors as wide spectrum of affected organs, non-specific symptoms or its rare occurrence (combined incidence of all types of amyloidosis is 10 in 1 000 000 inhabitants) makes amyloidosis difficult to be recognised in short time to render maximal treatment outcome. Its complex nature requires involvement of experienced clinicians from different specialisations such as nephrologists, cardiologists, haematologists or surgeons. The need for straightforward diagnostic workflow is especially pronounced for hereditary amyloidosis, where the mutation in one of the seven genes (transthyretin TTR, fibrinogen alpha FGA, apolipoprotein A1 APOA1, apolipoprotein A2 APOA2, lysozyme LYZ, gelsolin GSN or cystatin C CST3)1 4 leads to formation of amyloid that deposits in heart, liver, gastrointestinal tract, kidneys or nervous system (figure 1). The current diagnostic practice includes consecutive examination of coding regions of seven amyloidogenic genes by Sanger sequencing. The process either begins with sequencing of randomly selected gene or the most commonly affected gene (transthyretin) or the gene in which the mutation is linked with the damage of target organ where the amyloid accumulated. The first approach is laborious and expensive, while the latter one is patient specific and often leads to quick results. Hence, it requires decision of an expert with deep knowledge on amyloidosis, who is unfortunately not present in all medical centres.

Mutations causing hereditary amyloidoses sorted according to target organs or organ systems. CNS, central nervous system; GIT, gastrointestinal tract.
Incidence of hereditary amyloidosis is one case per million patients per year globally, but can vary in endemic areas. For example, transthyretin amyloidosis, the best known type of hereditary amyloidosis, illustrates that the incidences can vary worldwide, from one case in 538 people in an endemic area of northern Portugal6 to one case in 100 000 people in the US population.7 Penetrance may vary as well; in some cases penetrance can reach 100%, other cases may be incomplete and carriers of the gene may remain asymptomatic, yet their children can be affected clinically.8 In the Czech Republic, the incidence of amyloidosis is underachieved compared with other non-epidemic regions similar to the Czech Republic. We suppose that all types of amyloidosis are underdiagnosed or belatedly diagnosed and broader screening is needed.
Patients with hypertrophic cardiomyopathy (HCM) present heart impairment and therefore they meet one of the important, though non-specific, symptoms of amyloidosis. Cardiac amyloidosis is connected mainly to mutations in gene for transthyretin. Damage to the heart has been observed in 62% of patients with the developed disease.9 TTR gene can possess 85 heterozygous mutations in different exones.10 Other genes connected to heart impairment with lower incidence are FGA, APO AI and LYZ. Nevertheless, additional causal mutations still could exist, but remain undescribed.
Altogether, seven genes are responsible for hereditary amyloidosis and another four main genes are connected to other types of amyloidosis with not yet fully understood cause. A method that would allow straightforward, comprehensive and simultaneous testing of all amyloidogenic mutations is still lacking. Thus, we present results of newly designed amyloidosis panel that can be used for genetic testing and screening and should make the diagnostic process faster and more accurate. Presented approach can serve for large spectrum of medical institutions and laboratories on verification by Clinical Laboratory Improvement Amendments standards. We applied the panel on a cohort of patients with heart impairment, but it is suitable for patients with other organ dysfunctions suspected to be caused by amyloid deposits.
Patients and material/methods
Specification of cohort for hereditary amyloidosis screening
Randomly selected 40 patients with HCM served as a pilot cohort. This cohort size allowed optimal utilisation of sequencing chemistry. The samples of peripheral blood were collected between 2006 and 2015 from cardio centres in the Czech Republic. All patients were included in this study on signing informed consent. The study was conducted in accordance with the current version of Helsinki Declaration. The only clinical criteria for patient enrolment in the study were the presence of HCM of unknown origin (ie, patients negative for diabetes, hereditary cardiomyopathy, and so on) and parameters from magnetic resonance, ECG or echocardiography, which are known to be indicators of potential amyloid deposits. The patient’s baseline characteristics were as follows: male/female: 20/20 (50%/50%); median age of 55.4 years (range 27–80 years). 33.3% of patients had HCM positive familial history of HCM and 17.9% positive familial history associated with sudden death. Most of the patients had intermediate stage according to New York Heart Association system: 20.5-38.5-30.8-2.6-7.7% (I-II-III-IV-NA). Other clinical features are summarised in table 1.
Patient baseline characteristics
Sample collection and DNA isolation
A blinded cohort of 40 patients with HCM of unknown origin was analysed together with four positive and four negative control samples provided by the National Amyloidosis Centre London (UK) and tested previously by Sanger sequencing. DNA from non-coagulable peripheral blood stored in EDTA tube was isolated by MagCore automatised station (Anatolia Geneworks, Istanbul, Turkey). Quality and quantity of DNA were measured using Nanodrop 2000 Spectrophotometer (Thermo Scientific, Waltham, USA) and using Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, USA) (online supplementary table 1). Integrity of DNA samples was checked using 1% agarose gel electrophoresis.
Supplementary file 1
Next-generation sequencing design and library preparation
Capture probe set for amyloidosis panel library construction was designed with assistance of Agilent eArray tool (Agilent Technologies, Santa Clara, USA) (figure 2). Total genome part covered by probes was 63 kbp and spanned 11 genes (including introns and untranslated regions) involved in hereditary amyloidosis: TTR (HGNC:12405), FGA (HGNC:3661), APOA1 (HGNC:600), APOA2 (HGNC:601), LYZ (HGNC:6740), GSN (HGNC:4620), CST3 (HGNC:2475) or associated with other ‘amyloid’ diseases: PRNP (HGNC:9449), APP (HGNC:620), B2M (HGNC:914), ITM2B (HGNC:6174). For the detailed description and gene coordinates, see online supplementary table 2. The full design is available as .bed files on request. For the small-sized library, we selected HaloPlex Target Enrichment System (Agilent Technologies) which uses RNA-based probes and thus should provide better specificity for the capture. Libraries were prepared according to the manufacturer’s protocol from 225 ng of genomic DNA. Briefly, DNA was fragmented using enzymatic digestion with eight enzymes. Fragments were hybridised to the probe set in the presence of indexing primer cassette and the probe-fragment hybrids were captured on magnetic beads. Library of fragments was then amplified by PCR to produce a sequencing-ready, target-enriched sample.
Supplementary file 2

Example of a part of the panel design probes visualised in Integrative Genomics Viewer (IGV) program.
Fragmented DNA and final libraries were evaluated using Bioanalyzer 2100 (Agilent Technologies). For the examples of correctly and incorrectly prepared libraries, see online supplementary figure 1. Next-generation sequencing (NGS) was performed using MiSeq Reagent Kit V2—500 cycles (Illumina, San Diego, CA), expected coverage of sequencing was more than 200×.
Supplementary file 3
Bioinformatic analysis
Raw fastq reads were trimmed by Trimmomatic software11 and subsequently aligned to the ensembl GRCH38 reference genome by BWA MEM algorithm.12 Bam files were sorted and realigned by Picard Tools and GATK (http://picard.sourceforge.net).13 Mpileup format of alignments was reconstructed from sorted bam files by SAMtools.14 Variant calling format (VCF) files were generated by VarScan V.2.015 from mpileups (minimal coverage 10, minimal supporting reads 4, frequency threshold 0.3). VCFs were annotated against human reference genome GRCH38 with relevant GTF file (V.83) by our in-house scripts. Subsequently, all variants were compared with dbSNP (build 149) database (ncbi.nlm.nih.gov) and functional predictions of non-synonymous mutations were obtained from dbNSFP,16 MutTaster and Provean.17 18 Only non-synonymous single nucleotide variants (SNV) and INDELs in coding DNA sequence or splice sites have been further evaluated and visualised by Integrative Genomics Viewer browser.19 20
Validation by Sanger sequencing and restriction digest
If target genes were found mutated, confirmation of NGS results was performed with Sanger sequencing. Selected genes were amplified by PCR. Primers were designed to allow sequencing of desired polymorphism in a given gene. A list of primers is provided in online supplementary table 3. PCR was set up under the following conditions: 20 μL PCR reaction mix contained 0.5 U MyTaq polymerase, 1× MyTaq RED reaction buffer (both Bioline, UK), 8 µmol of each primer (Sigma Aldrich), PCR grade water and DNA template. Amplification was set up as follows: 95°C for 3 min, 35 cycles of 95°C for 30 s, 52°C for 30 s, 72°C for 90 s, followed by final extension 72°C for 10 min and constant incubation at 10°C. PCR products were separated by electrophoresis (5 V/cm) in 1% agarose gel and then purified using Gel/PCR DNA Fragments Extraction Kit (Geneaid, Taiwan) when positive result was obtained. PCR products were mixed with primers and sequenced in the company SeqMe, sro (Czech Republic). Each product was sequenced from both, forward and reverse, directions. Reads were visualised and assembled in Sequencher V.5.1 (Gene Codes, USA). To confirm the correct genotype in randomly chosen and ambiguous genotypes as well, restriction digest was used. The PCR products from above mentioned PCR reaction were digested by BsaAI enzyme (PRNP gene) and EagI enzyme (CST3 gene) (New England Biolabs). Restriction was set up under the following conditions: 15 µL final reaction mix contained 0.25 µL restriction enzyme, 1.5 µL 10× reaction buffer, PCR grade water and 0.5 µL PCR product. Reactions were incubated in 37°C for 60 min and products were checked by electrophoresis in 2.5% agarose gel.
Supplementary file 4
Statistical analysis
Frequencies of reference and alternative alleles were calculated from observed genotype frequencies. The obtained numbers were used to calculate expected genotype frequencies and the results were evaluated by Χ2 test to check whether our data correspond to Hardy-Weinberg principle.
Observed genotype frequencies were also compared with population frequencies available at 1000 Genomes Project21 and the differences were tested using Fisher’s exact test.
Sensitivity of NGS assay was calculated from Sanger and digestion data as the number of true positives given the sum of the number of true positives and false negatives. Specificity was calculated as the number of true negatives given the sum of the number of true negatives and false positives. The sensitivity and specificity values and CIs were calculated using online calculator MEDCALC.
Results
NGS metrics and SNVs summary
After processing of fastq files, the average number of mapped reads was 493 575 reads per patient and the average number of properly paired reads was 491 596 reads per patient. Given the size of the library (63 kbp) and the length of each read (250 bp), the average coverage was 1951 reads per site.
Four samples served as positive controls with mutations previously detected by Sanger sequencing in one of the following genes: APOA1 (p.Glu34Lys) and APOA1 (p.Leu60Arg), FGA (p.Glu526Val) and TTR (p.Val30Met). These variants were confirmed also by NGS using the presented workflow. In the tested cohort (40 patients), we detected amyloidogenic mutation (p.Glu82Lys) c.243G>A in TTR gene in one sample. In addition, we identified 12 different non-synonymous SNVs in seven genes; eight variants were shared in two or more patients (for details, see table 2 and online supplementary table 4).
Supplementary file 5
List of all captured variants in our cohort with genomic localisation, allele frequencies and risk prediction provided by program MutTaster
Validation of NGS data
Sanger sequencing and restriction digestion
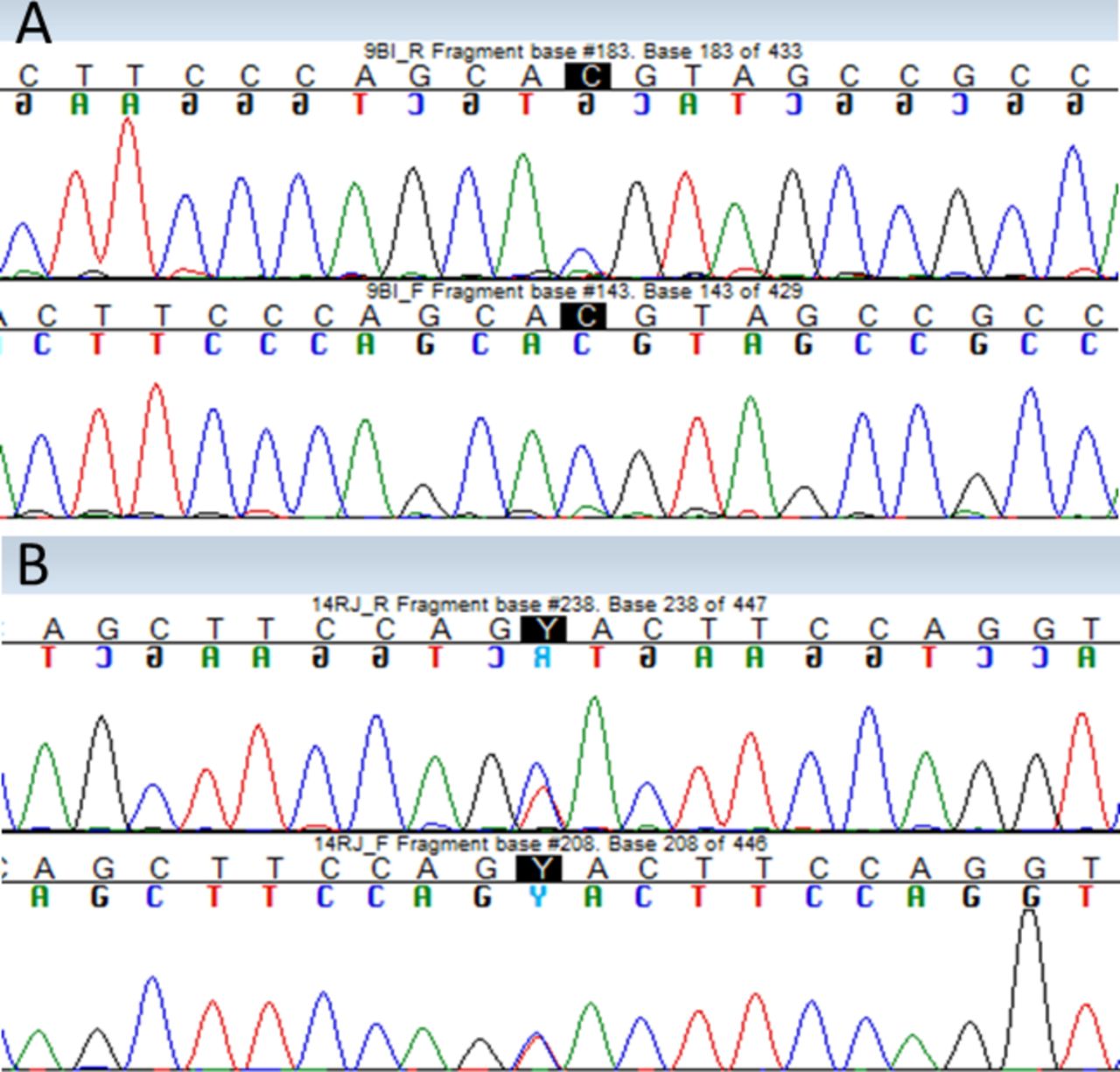
Ten cases of newly identified polymorphisms (in genes PRNP, CST3, FGA) were selected for subsequent validation by Sanger sequencing. The selection was made based on the proportion of homozygous and heterozygous variants in Illumina reads. We unambiguously verified the presence of the genotype suggested by NGS in 8 out of 10 tested cases. Example of Sanger sequencing confirmation is provided in figure 3. For the unconfirmed cases, we visually re-evaluated the alignment of Illumina reads and finally decided to use restriction enzyme digest to confirm the correct genotype. Restriction digestion confirmed results of NGS in one case.

{kind=link}
{kind=link}
![[jclinpath-2017-204978-SP3.jpg]](https://jcp.bmj.com/content/jclinpath/early/2018/04/28/jclinpath-2017-204978/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
Results of validation of next-generation sequencing (NGS) variants by Sanger sequencing. Verified bases are highlighted in black (A) homozygote variant PRNP gene Val129Met. (B) Heterozygous variant in FGA gene Thr331Ala.
The sensitivity of the NGS testing was 100.00% (95% CI 75.29% to 100.00%) and specificity was 93.33% (95% CI 68.05% to 99.83%).
Validation by allele frequency comparison
To prove quality of NGS data, frequency of nine non-amyloidogenic SNVs detected in our data set was compared with the frequency in Central European (CEU) population available at the SNPedia2 database. Calculated genotype frequencies were in agreement with observed genotype frequencies according to Χ2 test and this result allowed further testing of population frequencies by Fisher’s test. This test revealed no significant difference (P>0.05) of allele frequencies between both, tested and reference, populations (table 2), which indicate that our sequencing and variant calling approach was appropriate and provided reliable results.
Non-amyloidogenic variants candidate for functional tests
Frequency of three variants in our data set was very low and they also lacked a record in database of common polymorphisms. These facts, together with the effect predicted to be disease causing by at least one predictor (when available), suggest that the variants are potentially dangerous for the patient (table 2).
Insertion-deletion type of polymorphism was found only once for the entire data set. Specifically, the 24 nucleotide deletion was identified in PRNP gene coding region with unclear biological impact. However, this deletion spans octapeptide region important for the regulatory function of the mature protein.
We also captured two unique variants in one patient, (p.Glu599Lys) c.11795G>A (rs140304729) in APP gene together with (p.Leu18Pro) c.T>C]) in B2M gene. The latter mutation occurs in signal peptide of beta-2 microglobulin and its biological meaning is so far unknown, although its location in the signal peptide can affect protein targeting and secretion ability. Moreover, the Leu>Pro substitution heavily disturbs physical-chemical properties of the affected protein region with expectable biochemical consequences.
For the detailed overview of identified variants and their frequencies, see table 2.
Mutation causing hereditary amyloidosis
We revealed one positive sample for heterozygous mutation (p.Glu82Lys) in TTR gene. This mutation was first described by Briani et al in 201222 and the position is also annotated in Amyloidosis database (accessible at http://www.amyloidosismutations.com/).10
In our case, the patient was a 56-year-old woman with HCM of unknown significance, no history of neurological or cardiological disorders, who presented signs and symptoms of angina pectoris and dyspnoea. The patient has positive history for hypertension. Cardiac MRI findings were suggestive of amyloid deposition. Abdominal fat fine needle aspiration was positive. Endomyocardial biopsy was not accomplished because of high invasivity. No family history has proven. Descendants were recommended for genetic counselling. Patient characteristics were as follows: QTc interval 490 (ms), interventricular septum thickness 28 (mm), posterior wall thickness 13 (mm), left ventricle (LV) end-diastolic diameter 59 (mm), LV end-systolic diameter 35 (mm), left atrium end-systolic diameter 55 (mm), systolic blood pressure 140 (mm Hg), diastolic blood pressure 90 (mm Hg). Cardiac troponin I was increased (0.24 lg/L, normal values 0–0.15) as well as NT-proBNP (2385 ng/L, nv 0–900). Mutation was proven by Sanger sequencing.
Conclusion
Hereditary amyloidosis diagnostics in the Czech Republic
We aimed to increase detection potential of hereditary amyloidosis which seems to be underdiagnosed in the Czech Republic (population of approximately 10 million inhabitants). Disease incidence in neighbouring areas is 1 in 1 million inhabitants per year, but the real number of diagnosed cases in Czech Republic is only five cases. Genetic testing (using sequencing) should be accompanied by classical investigation methods for amyloid deposits such as imaging or biopsy followed by histological identification. For targeted screening of single genes in individual patients and for validation of NGS data as well, Sanger sequencing remains the gold standard method.
We designed a panel of 11 genes that covers all potential types of mutations causing amyloid deposits in various tissues. For the pilot experiment, we selected 40 patients with HCM of unknown origin plus positive and negative control samples for implementation of new panel for genetic testing and to uncover potential cardiac amyloidosis. The full-length gene coverage allows us to precisely examine all possible mutations responsible for disease manifestation and/or increase the chance of detection of a new variant. Our approach can be adopted by other researchers and clinicians and can be used for patients with wide spectrum of amyloidosis symptoms, not only heart impairment.
Reliability of NGS amyloidosis panel
We used for validation 14 SNVs for which we compared NGS results with one of the standard methods (Sanger sequencing or restriction digestion). Two cases of heterozygote alleles defined by NGS were not confirmed by Sanger. Subsequent testing by restriction digestion verified NGS results in one case and Sanger results in another case. This situation resulted in specificity values lower than 100% (specificity was 93.33% (95% CI 68.05% to 99.83%)). However, specificity of Sanger sequencing may also not always reach 100%, since this method has some limitations. On the other hand, NGS became a well-established method23 that is especially widely used in diagnostics of hereditary disorders and has been constantly improved in terms of base calling and variant calling accuracy. Specificity of NGS method in our study was 100%. Comparison of allele frequencies in our data set and in CEU population showed that our data correspond to general values and this fact yet supports the reliability of NGS method.
Altogether, our approach can be used for screening of large number of patients and all positive cases will be captured because of high sensitivity. Then positive cases should be retested by standard means and eventually some false positives, if present due to decreased specificity of NGS, will be discarded. Hence, all positive patients will be detected and will have chance to get the therapy.
True hereditary amyloidosis connected with mutation (p.Glu82Lys) in TTR gene
In this study, we discovered one patient with hereditary amyloidosis caused by mutation in TTR gene. The patient possessed several heart characteristics typical for amyloidosis. Examination by sequencing was necessary to confirm the nature and the origin of amyloid. Hereditary amyloidosis (late-onset ATTR) could be easily misdiagnosed, for example, (1) with the cardiac senile amyloidosis—wild-type ATTR amyloidosis,24 (2) when it is caused by de novo mutation (before they are deposited in public databases like Amyloidosis database10), or (3) due to its incomplete penetrance. In all cases, the treatment for patient would have been delayed.
Variant in TTR gene p.Glu82Lys is very rare and the case reported here represents only the second detection of the substitution on this position. We performed complete diagnostic workflow and the diagnosis was confirmed also by Sanger sequencing and by abdominal fat examination. Our data thus may serve as an independent confirmation of previously published data22 on this mutation.
Manifestation of genetic risks
Apart from detection of mutation causing hereditary amyloidosis, our data can show also some other risk alleles that are potentially dangerous for the patients. We identified several such polymorphic sites and tried to correlate them to clinical data that we obtained for the patients.
We detected one patient with eight amino acid deletion in the octapeptide region of PRNP protein. Deletions in octapeptide region might also play role in the inhibition of toxic effect of prion protein in prion pathologies.25 Similar deletion in octapeptide region might have relation to Alzheimer disease, although no significant correlation was ultimately proven.26
Also the variant p.Ala25Thr (g.[20:23637790_G>A]) of CST3 has been associated with increased genetic predisposition of Alzheimer disease and age-related macular degeneration,27 but we did not observe that the risk manifested in our cohort, most probably due to relatively young examined patients.
FGA polymorphism predicts increased risk of venous thromboembolism or early pregnancy loss.28 29 In our cohort, we identified two patients with diagnosed thromboembolism and both of them possessed the risk genotype.
Potential novel causal mutations: further studies needed (B2M p.Leu18Pro and GSN)
The effect of the variant p.Leu18Pro (g.[15:44711599_T>C]) in beta-2 microglobulin occurs in signal peptide region of protein and has not been studied yet, thus we can only hypothesise about its potential impact. Amino acid substitution to proline often alters the structure of the protein and leads to formation of beta sheets. Such change in other protein can affect protein localisation and secretion,30 31 moreover the similar conformation change is also typical for amyloid-forming proteins. The patient bearing this variant in our cohort evinced the highest blood pressure (systolic/diastolic 179/95), therefore we can speculate about the effect of the mutation also on this parameter. Indeed, increased beta-2 microglobulin serum levels were previously associated with hypertension.32
As this is the only variant not occurring in the database of known polymorphisms (dbSNP) and has predicted deleterious effect (MutTaster, Provean), it represents the best candidate for more detailed description of the effect of this variant, further association studies or functional tests.
We detected also four variants in GSN gene: p.Thr563Ser, p.Thr565Met, p.Ala78Thr and p.Trp14Arg. Population frequencies of these variants are very low in normal population, while they are slightly higher in our data set, yet statistically not significant. The sense of these mutations is unknown at this time but we hypothesise some of them (p.Thr563Ser, p.Ala78Thr) could have a damaging effect according to the functional predictors (table 2).
Benefits of our approach
Rare hereditary diagnoses are especially suitable for screening by NGS. It offers examination of larger genomic region compared with Sanger sequencing. Tens of genes can be sequenced at time for significantly less money and with less effort including the process of final data analysis. We prepared a gene panel appropriate for hereditary amyloidosis that can be adopted by other laboratories. We performed a number of controls of the sequencing process and together with current progress of NGS introduction in cancer research we show that NGS method can be readily introduced and validated for diagnostic purposes. Improved diagnostic potential will help the patient to get treatment in shortest possible time.
Take home messages
Hereditary amyloidosis represents a rare monogenic disease caused by germline mutations in 11 amyloidogenic precursor protein genes.
Targeted genetic screening using next-generation sequencing (NGS) panel can facilitate diagnostic process of rare hereditary diseases.
We detected one case of hereditary amyloidosis among patients with hypertrophic cardiomyopathy of unknown significance using NGS.
Our NGS data suggest possible presence of new causal mutations of hereditary amyloidosis.
Acknowledgments
We are also grateful to Dr Fedor Kyukov and Dr Elena Kryukova who moderated this paper and in that line improved the manuscript significantly.
References
Footnotes
Handling editor Mary Frances McMullin.
Contributors ZKC, TS and KG performed the experiments. PV performed the bioinformatical analysis. ZKC, TS, PV, JJ and AB wrote the manuscript. ZKC, TS, JJ and RH designed the study. JJ, AB and PVa provided the clinical and samples background. JF, TJ, MH and RH improved and proofread the manuscript.
Funding This work has been realised with the support of the IRP03_2018_2020, MH CZ-DRO-FNOs/2016 and with the support by the Ministry of Health 15-29667A project: Strengthening international cooperation in science, research and education 01211/2016/RRC and Ministry of School and Education (LO1304) and Specific university research of the Faculty of Medicine (University of Ostrava) project numbers SGS 02/LF/2014-2015, SGS 09/LF/2016-2017. The project was supported by Amgen.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics Committee of University Hospital Ostrava.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement BAM files are available at the corresponding author upon request.
Correction notice This article has been corrected since it was published Online First. The sentence ’Specifically, the 25 nucleotide insertion was identified in PRNP gene coding region…' on page 4 has been corrected. The mutation "p.Glu62Lys" has been correct throughout the paper.