Article Text

Abstract
Cyclin-dependent kinase 12 (CDK12) belongs to the cyclin-dependent kinase (CDK) family of serine/threonine protein kinases that regulate transcriptional and post-transcriptional processes, thereby modulating multiple cellular functions. Early studies characterised CDK12 as a transcriptional CDK that complexes with cyclin K to mediate gene transcription by phosphorylating RNA polymerase II. CDK12 has been demonstrated to specifically upregulate the expression of genes involved in response to DNA damage, stress and heat shock. More recent studies have implicated CDK12 in regulating mRNA splicing, 3’ end processing, pre-replication complex assembly and genomic stability during embryonic development. Genomic alterations in CDK12 have been detected in oesophageal, stomach, breast, endometrial, uterine, ovarian, bladder, colorectal and pancreatic cancers, ranging from 5% to 15% of sequenced cases. An increasing number of studies point to CDK12 inhibition as an effective strategy to inhibit tumour growth, and synthetic lethal interactions have been described with MYC, EWS/FLI and PARP/CHK1 inhibition. Herein, we discuss the present literature on CDK12 in cell function and human cancer, highlighting important roles for CDK12 as a clinical biomarker for treatment response and potential as an effective therapeutic target.
- cancer genetics
- cell biology
- C-MYC oncogene
- breast cancer
- ovarian cancer
Statistics from Altmetric.com
Introduction
CDK12 (cyclin-dependent kinase 12; CRKRS, CRKR or CRK7) was first identified from cDNA screens for cell cycle regulators related to cdc2 kinases.1 In contrast to cyclin-dependent kinases (CDKs) that promote transition between different phases of the cell cycle, CDK12 is a transcriptional CDK with specific roles in regulating transcription of genes involved in cellular responses to DNA damage and stress.2–4 Emerging studies continue to dissect the role of CDK12 in cell function and cancer and have illuminated its potential clinical use as a biomarker and therapeutic target.
Structure and expression
On chromosome 17q12, the CDK12 gene encodes a 1490 amino acid protein with a molecular weight of 164 kDa.5 The closely related CDK13 (located on 7p14) shares 43% sequence homology and a largely conserved kinase domain (KD) (figure 1). The central KD mediates phosphorylation of RNA Pol II,6 consisting of ~300 amino acids that shares 42% identity to human cdc2 and featuring characteristic analogous threonine and tyrosine residues required for cdc2 inactivation.5

Schematic structures of the CDK12 and CDK13 genes and chromosomal location of the respective genes. CDK, cyclin-dependent kinase; KD, kinase domain; PRM, proline-rich motifs; RS, arginine/serine rich motifs.
Arginine/serine-rich (RS) motifs are critical components of proteins involved in pre-mRNA processing and can function as a nuclear localization signal.7 CDK12 contains 21 RS motifs within the first 400 amino acids.5 Proline-rich motifs are found between the RS domain and central KD as well as at the C-terminal region (figure 1). These regions contain consensus binding sites for SRC Homology 3 and WW domains, indicating potential protein interaction partners from a wide range of signalling pathways.8 9 A unique C-terminal helix outside the canonical kinase fold of CDK12 facilitates its interaction with cyclin K.10 11 The flexibility of this C-terminal extension was found to be critical for the kinase and ATP-binding activity of CDK12 and has directed the development of novel inhibitors to CDK12/13.12 13
CDK12 is ubiquitously expressed, as demonstrated by human tissue northern blots in a panel of RNAs from different human tissues.5 RNA sequencing analysis and immunohistochemical staining of 95 human individuals representing 27 tissue types also detected CDK12 in all tested tissues.14 Compared with other tissues, higher CDK12 expression was generally detected in male and female reproductive tissues, endocrine tissues, bone marrow, spleen and lymph nodes. Staining for CDK12 expression was also mainly localised to the nucleus, as suggested by its RS motifs and cellular functions.14
Functions
CDK12 functions as a complex with cyclin K, with its most well-characterised roles in the regulation of gene transcription.3 The strong functional link between CDK12 and cyclin K is reflected in the fact that knockdown of either protein results in similar phenotypes and affected genes, leading to genomic instability.3
Transcription, mRNA processing and the DNA damage response
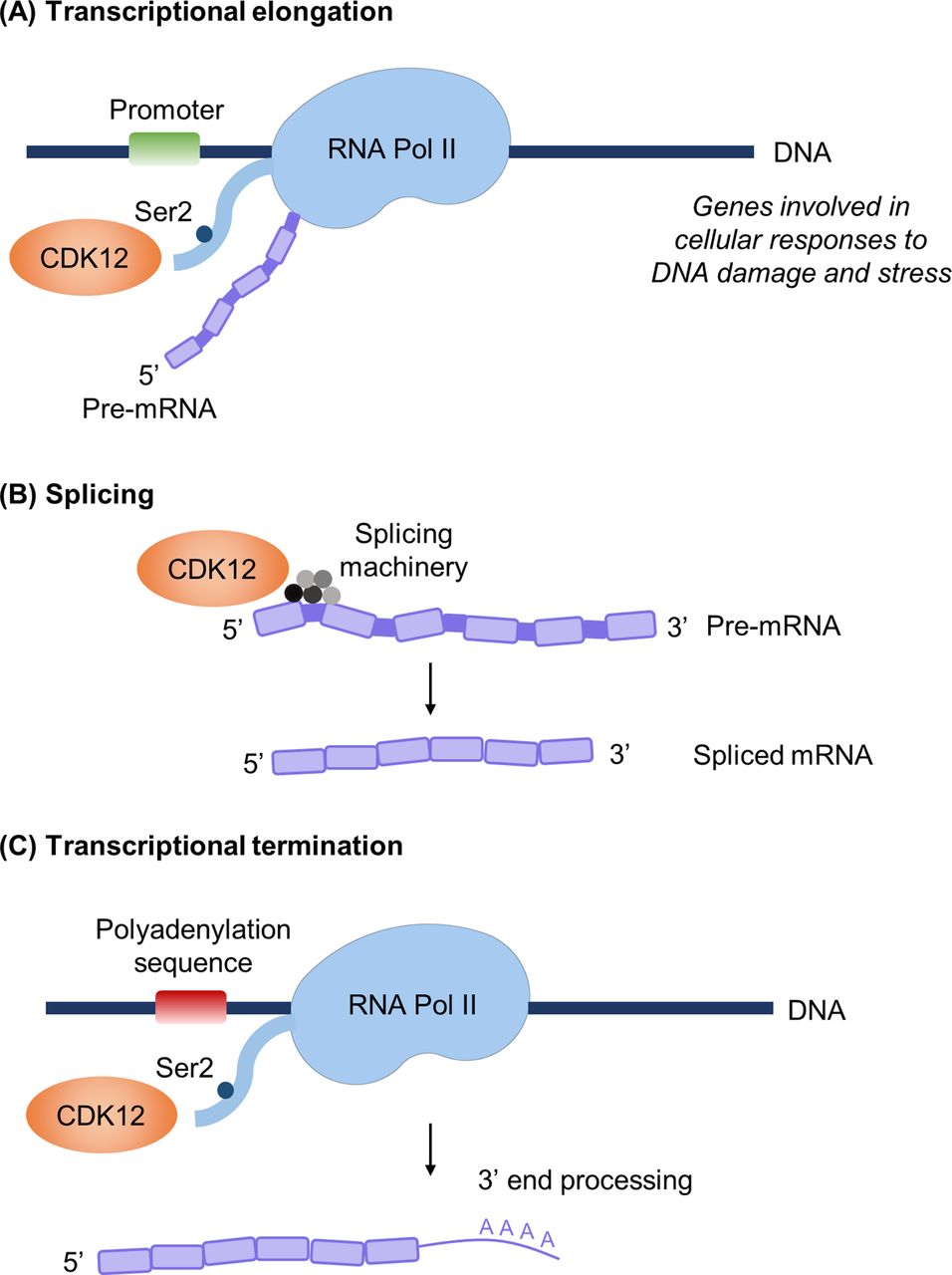
The CDK12/cyclin K complex phosphorylates RNA Pol II at Ser2 (Ser2p-RNA Pol II), which is thought to be a critical step in transition from transcriptional initiation to elongation3 6 15 16 (figure 2A). In vitro, CDK12/cyclin K was also shown to phosphorylate Ser5 of RNA Pol II, suggesting potential regulation of transcription initiation.11 In cells depleted of CDK12, expression microarrays demonstrated that only 2.67% of tested microarray genes were altered.3 Moreover, of the genes altered, the majority were downregulation of genes with large numbers of exons. Gene classification revealed an enrichment of genes involved in DNA replication, recombination and repair centred on the BRCA1 module, and cells with knockdown of CDK12 had significantly lower levels of BRCA1, ATR, FANCI and FANCD2.3 CDK12 or cyclin K knockdown sensitised cells to DNA-damaging agents,3 suggesting that CDK12/cyclin K is a master regulator of proteins specifically involved in DNA damage repair (DDR) and response to DNA damage. CDK12 was also reportedly required for function of CncC, the Drosophila homolog of the stress-activated Nrf2 transcription factor, and expression of oxidative stress response genes, but not that of general housekeeping genes or cell viability.4 Collectively, these studies indicate that CDK12 regulates specific subsets of genes involved in cellular responses to DNA damage, stress and heat shock.3 4 6

Known functions of CDK12. (A) CDK12 phosphorylates RNA polymerase II (RNA Pol II) at Ser2, which promotes transcriptional elongation. (B) CDK12 interacts with RNA-processing factors to regulate splicing. (C) CDK12-mediated phosphorylation of RNA Pol II couples transcription and mRNA 3’ end processing. CDK12 reportedly regulates the expression of a distinct subset of genes, including those involved in the DNA damage response, cellular stress and heat shock. CDK, cyclin-dependent kinase.
The characteristic RS motifs of CDK12 strongly indicate functions in pre-mRNA processing.5 Splicing factors are thought to be stored in subnuclear structures known as nuclear speckles,17 and CDK12 localises to nuclear speckles and spliceosome components.5 Indeed, mass spectrometry of CDK12-associated proteins identified a strong enrichment for RNA-processing factors18 and an enrichment of genes involved in RNA splicing machinery19 (figure 2B). These studies also showed that CDK12 regulates the expression and alternative last exon (ALE) splicing of genes with long transcripts and large numbers of exons.19 CDK12 can also indirectly regulate RNA processing by regulating phospho-epitopes on the C-terminal domain of RNA Pol II.18
Ser2p-RNA Pol II couples transcription and mRNA 3’ end processing by interacting with polyadenylation and termination machinery at the 3’ ends of mRNA.20 Davidson et al demonstrated that CDK12-mediated phosphorylation of Ser2p-RNA Pol II recruits the cleavage and polyadenylation factor CstF77 to ensure efficient 3’ end formation21 (figure 2C). CDK12 was demonstrated to be required for optimal pre-mRNA processing of the MYC gene, with gene depletion reducing levels of polyadenylated MYC RNA.21
DNA replication
A recent study demonstrated that the CDK12/cyclin K complex is required for mammalian cell proliferation.22 Specifically, CDK12/cyclin K mediates phosphorylation of cyclin E1 at Ser366, which blocks interaction with its binding partner, CDK2, during pre-replicative complex assembly in early G1 phase.22 Although cyclin E/CDK2 normally accumulates at the G1/S transition to promote S phase entry, aberrant cyclin E1/CDK2 activity in early G1 has been shown to inhibit pre-replicative complex formation.23 Regulation of efficient pre-replication complex assembly by CDK12/cyclin K suggests novel roles in mediating crosstalk between DNA replication and gene transcription.22
Development
CDK12 expression is critical in mouse embryonic development. In vivo, Cdk12 activity was found to be critical from stage E3.5, and Cdk12 deficiency leads to arrest and embryonic lethality by stage E6.5.24 Cdk12 -/- embryos grown in vitro display spontaneous DNA damage and reduced expression of DNA damage response genes, including Atr, Brca1, Fanci and Fancd2.24 CDK12/13 and cyclin K are also required for self-renewal in embryonic stem cells, with knockdown of these proteins leading to differentiation.25 Roles in proper development of neural cells have been described, with mice expressing conditional deletion of Cdk12 in neural progenitor cells (NPCs) dying shortly after birth and exhibiting microcephaly. NPCs of these mutant mice also displayed lower expression of DDR genes, increased double-strand breaks and increased apoptosis.26 These findings recapitulate critical roles for CDK12 in maintaining genomic stability and expression of DDR genes in development.
CDK12 and CDK13 are known to share similar biological processes, with both regulating RNA splicing and alternative splicing by virtue of their RS motifs,27–29 maintaining self-renewal in mouse embryonic stem cells25 and regulating axonal elongation.30 Despite their similarity in sequence and interaction with cyclin K, CDK12 and CDK13 do not have identical functions and are likely to have evolved separately from a common ancestor gene.30 Recent studies indicate that CDK13 regulates distinct subsets of genes and biological processes from CDK12, including snRNA and snoRNA gene expression,31 and extracellular/growth signalling pathways.32
Cdk12 mutations in human tumours
Analysis of the CDK12 gene across The Cancer Genome Atlas (TCGA) revealed mutations, amplifications or deep deletions in 30/32 tumour types (figure 3). Tumour types with the highest percentage of cases with aberrant CDK12 (ranging from 5% to 15% of sequenced cases) include oesophageal, breast, endometrial, uterine and bladder carcinomas. Stomach, colorectal, pancreatic ductal and ovarian serous adenocarcinomas also feature significant levels of aberrations.
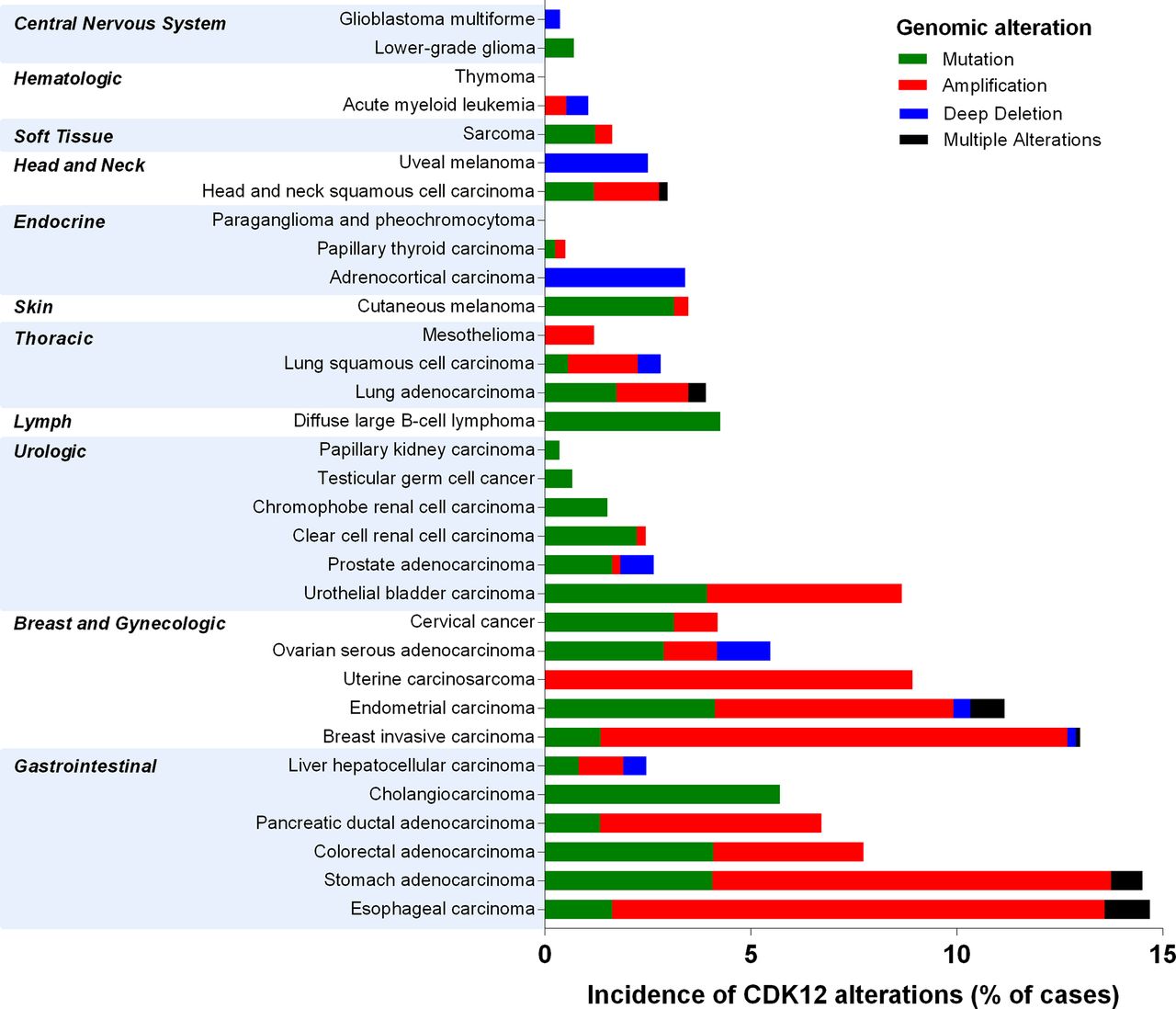
Genomic alterations of the CDK12 gene across TCGA. Data downloaded from TCGA Provisional data sets on cBioportal (http://www.cbioportal.org/) in May 2018. CDK, cyclin-dependent kinase; TCGA, The Cancer Genome Atlas.
The functional roles of CDK12 gene mutations, amplifications, deletions or variations in its expression remain incompletely understood, with both tumour-suppressive and tumorigenic roles proposed for CDK12. As far as its tumour-suppressive roles, two studies33 34 have reported that the majority of CDK12 mutations in high-grade serous ovarian carcinoma (HGSOC) are homozygous point mutations in the KD that abrogate the catalytic activity of CDK12. Loss-of-function of CDK12 results in decreased homologous recombination and enhanced sensitivity to DNA cross-linking agents and poly (ADP-ribose) polymerase (PARP) inhibitors.
Inactivation of the CDK12 gene has been associated with a unique genomic instability pattern characterised by up to 800 tandem duplications (TD; up to 10 Mb in size) per tumour that were quasi-randomly distributed along the genome.35 Denoted the CDK12 TD-plus phenotype, these TDs affected over 10% of the genome and were observed in 4% of serous ovarian carcinomas and up to 2% of prostate adenocarcinomas.35
On chromosome 17, the CDK12 gene is located approximately 200 kb proximal to the HER2 (ERBB2) oncogene and is frequently coamplified in breast tumours.36 37 In cohorts of primary breast cancer, high CDK12 expression correlated with HER2 status, suggesting oncogenic roles for CDK12 in this context.38 Tumorigenic roles for CDK12 were recently proposed through ALE splicing of DNAJB6, a HSP40 family chaperone, which promoted cell invasion and migration in HER2-amplified breast tumour cells.19 Other studies reported that 13% of HER2-positive breast cancers showed out-of-frame rearrangements of CDK12 resultant from the amplification breakpoint in the HER2 amplicon converging on CDK12.39 This led to decreases in CDK12 expression, loss-of-function and sensitivity to PARP inhibitors.39 In HER2-amplified MKN7 gastric cancer cells, gene fusions involving CDK12 and HER2 were reported.40 These fusion transcripts were predicted to result in truncation of CDK12 protein, but were not in-frame to HER2. CDK12 mutations have also been reported in non-small cell lung cancer,41 lung adenocarcinoma,42 follicular lymphoma,43 oesophagogastric tumours44 and advanced carcinoma of unknown primary.45
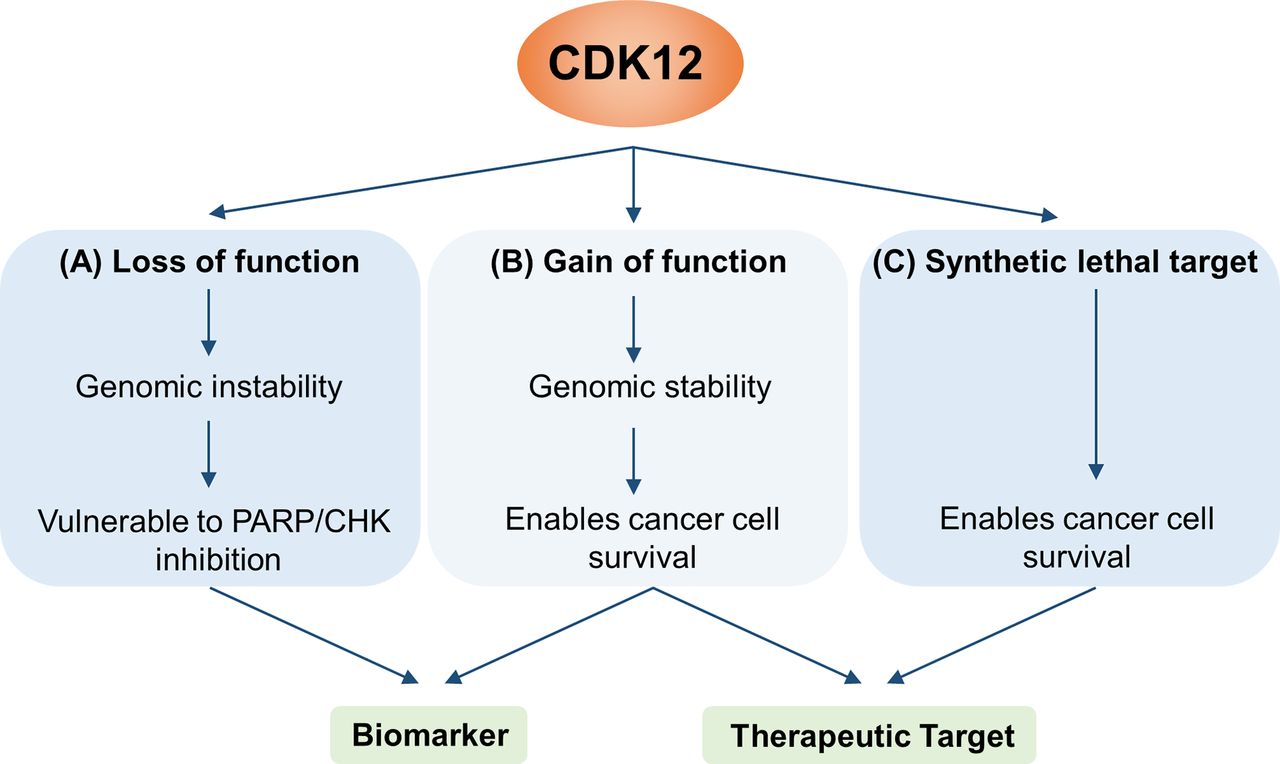
It is clear from these studies that the functional implications of CDK12 mutations are case-dependent and context-dependent. Continued elucidation of the specific roles of CDK12 will be important for its use as a biomarker to inform patient stratification for therapeutic intervention38 (figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Potential for clinical use of CDK12 as a biomarker and/or therapeutic target. (A) CDK12 mutations that confer loss of function have been reported to promote genomic instability, rendering cancer cells more susceptible to PARP/CHK inhibitors. (B) On the other hand, CDK12 mutations that cause gain of function (eg, amplification) could theoretically potentiate cancer cell survival by promoting expression of DNA damage repair genes. Though there are currently few reports of this, such a situation would enable use of CDK12 as a biomarker of drug response/clinical outcome or as a drug target. (C) In cases where CDK12 is not necessarily mutated, CDK12 can enable tumour progression driven by genes such as MYC and EWS/FLI. These synthetic lethal interactions also provide an opportunity for therapeutic targeting. CDK, cyclin-dependent kinase.
Cdk12 as a therapeutic target: SYNTHETIC lethal partners in the context of cancer
In addition to its potential role as a clinical biomarker, recent studies have highlighted CDK12 as a therapeutic target for cancer (figure 4). Inhibition of transcriptional CDKs could be an effective strategy to overcome resistance to targeted therapies, including erlotinib and crizotinib.46 Numerous other studies have identified specific genetic or cellular contexts that confer enhanced sensitivity to CDK12 inhibition, including MYC dependency, PARP inhibition and EWS/FLI rearrangement.
MYC
MYC is a global transcription factor and central driver of many human cancers, which has proven to be difficult to inhibit directly.47 48 To discover synthetic lethal genes with the MYC oncogene in an isogenic setting, we performed a siRNA screen using human fibroblasts with overexpression of cMYC.49 In this study, we first reported CDK12 as synthetic lethal with cMYC. These findings were corroborated in an independent study demonstrating that CDK inhibition triggered massive downregulation of cMYC expression and its related genes.50
Among the top genes identified as synthetic lethal with MYC were genes that regulate RNA polymerase II and cell cycle checkpoint control, including GTF2H4, POLR2E, RAD21 and WEE1.51 Additionally, deregulated MYC is known to induce replicative stress by accelerating the rate of DNA replication, pointing to replication-coupled DDR as a targetable weakness in MYC-driven tumours.52 53 The overlap between this MYC signature and the known cellular functions of CDK12 as well as the requirement of CDK12 for optimal processing of cMYC,21 collectively indicate CDK12 could be an effective therapeutic target for MYC-dependent cancers.
PARP and CHK1 inhibitors
A genome-wide PARP1/2 inhibitor screen identified CDK12 as a sensitiser to olaparib and found CDK12 mutations were a clinically relevant biomarker of PARP1/2 inhibitor sensitivity in HGSOC.54 Moreover, most HGSOC cases displaying CDK12 mutations were mutually exclusive with BRCA1/2 mutations, suggesting cells can use one of multiple strategies to achieve similar phenotypes. Supporting this, primary and acquired resistance to PARP inhibitors could be overcome by CDK12 inhibition in BRCA wild-type and mutated models of triple negative breast cancer.55 This has led to clinical trials exploring this combination in advanced solid tumours (NCT01434316). Recently, Paculová et al proposed that CDK12-deficient or BRCA1-deficient cells are reliant on the downstream S phase checkpoint kinase CHK1 for survival.56 Loss of CDK12 or BRCA1 was found to potentiate the antitumour activity of CHK1 inhibitors irrespective of p53 status.56
EWS/FLI
Ewing sarcoma tumours are characterised by chromosomal rearrangements resulting in the fusion protein EWS/FLI, a potent transcriptional activator and transforming gene in this disease.57 A recent study reported that inhibition of CDK12 was specifically responsible for synthetic lethality in Ewing sarcoma cells with EWS/FLI rearrangement.58 Treatment of these cells with the specific CDK12/13 inhibitor, THZ531, preferentially repressed expression of DDR genes and was synergistic with PARP inhibitors. Interestingly, CDK12 is rarely mutated in Ewing sarcoma tumours (TCGA), suggesting that mutations in CDK12 are not necessary to confer its role as an effective therapeutic target.
Tumours driven by oncogenes such as MYC and EWS/FLI are highly dependent on transcriptional programmes that converge on RNA Pol II59 60 and the need for DDR gene expression to facilitate rapid replication.61 Thus, impairing the function of CDK12 as both a transcriptional coactivator and specific regulator of DNA damage related proteins could explain the synthetic lethal interactions described above, representing a promising therapeutic strategy for these cancer types.
Take home messages
Cyclin-dependent kinase 12 (CDK12) complexes with cyclin K to regulate transcriptional elongation, mRNA processing, proliferation and development. It regulates specific subsets of genes involved in cellular responses to stress, heat shock and DNA damage.
Genomic alterations in CDK12 are frequently observed in human cancers. In high-grade serous ovarian carcinoma, HER2-positive breast cancer and lung adenocarcinoma, loss-of-function mutations have been reported, which decrease homologous recombination and enhance sensitivity to chemotherapy and PARP inhibitors. In contrast, CDK12 gene amplification could contribute to cancer fitness by constitutive engagement of DNA repair pathways.
Synthetic lethal interactions have been reported for CDK12 with MYC, EWS/FLI and PARP inhibitors and have led to growing interest as a therapeutic target and biomarker for response in cancer treatment.
Acknowledgments
The authors thank members of the Kemp laboratory for helpful discussions.
References
Footnotes
Handling editor Des Richardson.
Contributors GYLL wrote the manuscript and prepared all figures. CG and CJK contributed to writing of the manuscript.
Funding GYLL is funded by the Ovarian Cancer Research Fund Alliance. CJK is funded by grants from the NIH/NCI (U01 CA217883, U54 CA132381).
Competing interests CG and CJK are cofounders of and have ownership interests in SEngine Precision Medicine. GYLL has no competing interests to declare.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.