Article Text

Statistics from Altmetric.com
Introduction
Since October 2020, the emergence of novel lineages of SARS-CoV-2 is generating widespread concern.1 They are characterised by distinct genetic features that might lead to increased transmissibility and/or immune evasion from previous infection or vaccination.2 Therefore, genomic surveillance gains and increasingly important role to track the emergence, the spread and the transmissibility of these lineages.3 Our unpublished data demonstrated the fast spread of the B.1.1.7 ((WHO)-designated variant of concern Alpha) lineage in our territory, reaching the 100% prevalence in about 2 months.
The University Hospital of Udine is the hub that serves the former province of Udine (Italy) and, between April and May 2021, it processed 97 000 nasopharyngeal swabs. Specimens resulted positive with standard molecular biology techniques (Allplex SARS-CoV-2 Assay by Seegene) were analysed by high-resolution melting (HRM) to evaluate the presence of specific variants on the Spike glycoprotein (p.E484K/Q, p.N501Y, p.L452R, p.K417N/T, p.P681H) considered key features of almost all the novel identified lineages and/or associated with the lack of response to monoclonal antibodies and convalescent plasma treatments.4 About 370 positive samples with Ct values ≤28 (3.81% of total positive samples) were randomly selected and subsequently underwent next-generation sequencing (NGS) to establish their belonging lineage, in order to manage local public health responses and reorganise hospitalisation. Amplicon-based sequencing was performed on an Ion GeneStudio S5 system (Thermo Fisher Scientific). Sequences kept for further investigations were the ones possessing a mean depth of coverage ≥500 and a percentage of gaps≤20% of the entire sequence. For variant calling, variants with a genotype quality score ≥30, a coverage (flow total depth at position (FDP)) ≥500 and a minimum alternate allele frequency of 70% (≥70%) were kept for further investigations. FASTA sequences were analysed with the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) web tool developed by the Centre for Genomic Pathogen Surveillance (https://pangolin.cog-uk.io/) and the Ultrafast Sample placement on Existing tRee (UShER) web tool by the University of California Santa Cruz (UCSC) genome browser (https://genome.ucsc.edu/cgi-bin/hgPhyloPlace).
We identified 13 samples displaying an uncommon genotype, classified as B.1.621. It originates from the B.1 lineage and the shared genomic features harboured by our cluster are enlisted in table 1.
List of shared mutations of the SARS-COV-2 B.1.621 lineage isolated in the North East of Italy
Ten out of 13 sequences have been shared in the GISAID database (EPI_ISL_2613608, EPI_ISL_2613609, EPI_ISL_2613606, EPI_ISL_2613607, EPI_ISL_2613611, EPI_ISL_2613610, EPI_ISL_2613604, EPI_ISL_2613605, EPI_ISL_2613602, EPI_ISL_2613603). The remaining three were omitted due to quality criteria, although B.1.621 assignment was confirmed by both PANGOLIN and UShER online tools. As evidenced in figure 1A, our samples overlap with the other B.1.621 sequences in Nextclade V.2.29.1web application (https://clades.nextstrain.org/). To better characterise whether the 10 genomes analysed displayed some differences, we performed a phylogenetic analysis with Molecular Evolutionary Genetics Analysis V.11 using the maximum Likelihood method (Tamura-Nei method). Although few, samples cluster into two main subgroups (figure 1B).
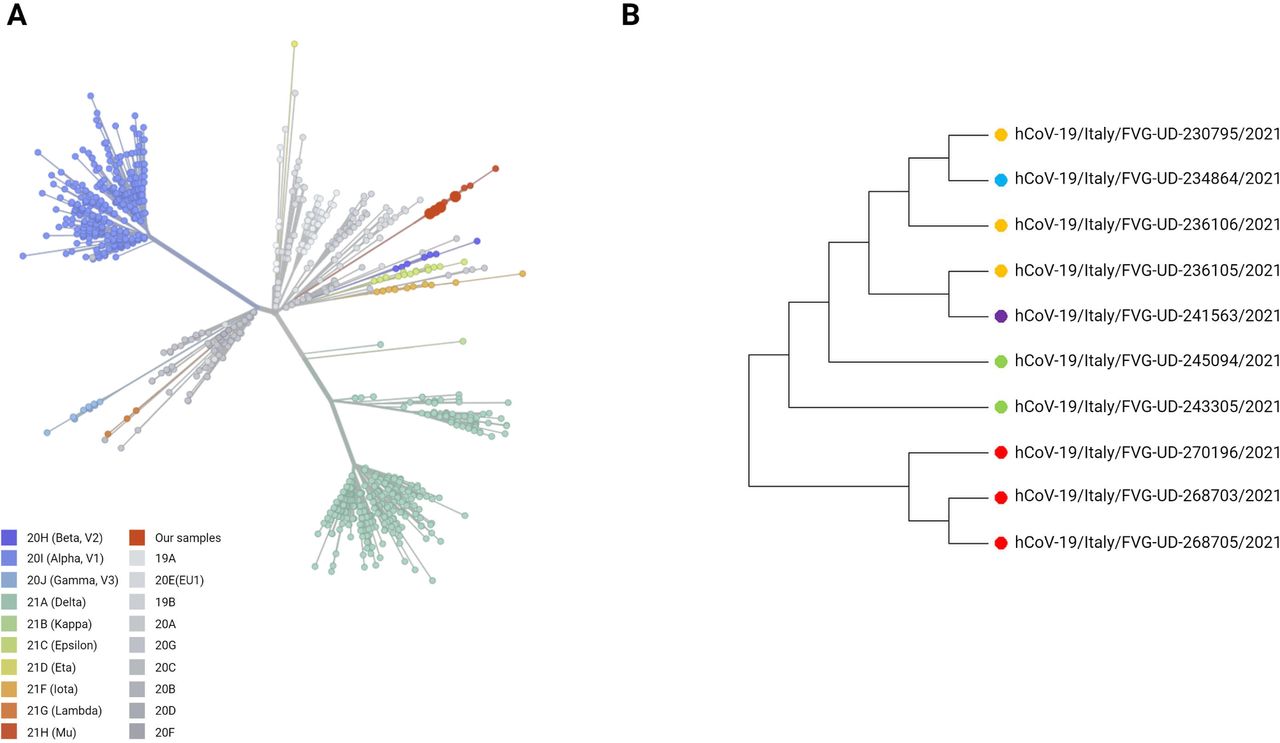
Phylogenetic analysis of B.1.621 samples. (A) Clades distribution according to the Nextclade online tool. Our sequences are represented as big red dots. (B) Phylogenetic tree of the 10 B.1.621 sequences based on the maximum likelihood method of MEGA V.11. Created with Biorender.com. FVG, Friuli Venezia Giulia; MEGA, Molecular Evolutionary Genetics Analysis.
Since epidemiological or in vitro evidences on the impact of this lineage on transmissibility, severity and/or immunity are by far preliminary, we promptly traced subjects who came into contact with the members of our cohort to prevent a putative outbreak and to avoid the possible spread of this lineage in our territory.
Indeed, this lineage has been identified in early January 2021 in Colombia,5 and it has been associated to sporadic infections due to contacts with travellers. It has been enlisted within the seven variants of interest by the European Centre for Disease Prevention and Control), in the ‘Situation updates on COVID-19’ document of 24 June.6 Furthermore, 624 sequences referring to this lineage have been deposited in the PANGOLIN database (https://cov-lineages.org/lineages/lineage_B.1.621.html). Our data document the first cluster of B.1.621 infections in Italy.
Given the rapid spread and the ability of viruses harbouring the p.E484K to escape from neutralising antibodies, this study emphasise the importance of monitoring the circulation of this strain and other variants of interest.2 Indeed, HRM-based screening is mandatory to early identify uncommon genotypes within the area of interest in order to both forewarn local healthcare facilities and prioritise NGS sequencing.7
Noteworthy, since January 2021, our approach allowed us to identify subjects infected by diverse lineages, such as B.1, B.1.1.7, B.1.177, B.1.221, B.1.258, B.1.1.317, B.1.1.318, C.17 (unpublished data). Focusing on data belonging to the timespan covered by this study, four major lineages were present in the ex-province of Udine. It is likely to assume that the occurrence of the lineages with lower frequency in our territory is attributable to international travels. These data are summarised in figure 2.

{kind=link}
{kind=link}
Lineage distribution across samples underwent next-generation sequencing between April and May 2021 in Friuli Venezia Giulia. B.1.177: 0.55%; B.1.258: 0.27%; P.1: 0.55%; C.36.3: 1.64%; B.1.620: 0.27%; B.1.621: 3.55%; B.1.1.7: 93.17%.
Discussion
In this analysis, we highlighted the local occurrence of SARS-CoV-2 lineages in the former province of Udine (Italy). Since March 2021, the prevalent variant in Friuli Venezia Giulia is the Alpha one, known to have a substantial transmission advantage over other lineages.8 Nonetheless, we witnessed the occurrence of a small percentage of variants of interest or variants under monitoring, mostly introduced by travellers. In this subgroup, the main lineage is represented by the B.1.621, being the 62.5% of the non-alpha ones.
The cluster we were able to track was mostly composed by individuals of South American origin (ie, Bolivia, Argentina and Colombia). Only one patient was hospitalised due to COVID-19 but discharged after 2 days with a diagnosis of mild pneumonia, the remaining being paucisymptomatic.
Notwithstanding, this report is flawed by the fact that a disclosure of interpersonal contacts was made on a voluntary basis, and therefore, the information may not be entirely reliable, hence, additional instances of transmission might have been missed.
Despite limitations, these findings have serious implications for public health agencies responding to SARS-CoV-2 variants of concern. Our data show that prompt contact tracing of confirmed cases and extensive collection of nasal swabs from close contacts, coupled to isolation or quarantine for SARS-CoV-2 infected subjects, can be effective in extinguishing local outbreaks. Genetic surveillance programmes must be indeed improved and implemented for the rapid detection and tracking of novel lineages, that is, B.1.621, in order to control their spread and alert public health authorities.
Ethics statements
Patient consent for publication
Ethics approval
Ethical approval was obtained from the Medical Research Ethics Committee of the Region Friuli Venezia Giulia, Italy (Consent CEUR-2020-Os-033).
Footnotes
CM and CDS are joint first authors.
Handling editor Tahir S Pillay.
CM and CDS contributed equally.
Contributors CM and CDS contributed equally to this work. CM and CDS: concept, design, testing, analysis and manuscript writing; SM and CP: concept, design, editing; ES and CT: concept, collection of clinical data and supervision; GD and FC: editing and supervision. All authors gave their final approval for this version of the article to be published.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.