Article Text

Abstract
Homocystinuria (HCU) refers to a group of inherited disorders of homocysteine metabolism associated with high blood homocysteine concentration, thromboembolic tendency and neurocognitive symptoms. The most common causes of a high blood homocysteine relate to underlying vitamin B12 or folate deficiency which must be excluded first. Thereafter, an inherited metabolic condition can be considered.
The most prevalent inherited disorder of homocysteine metabolism is classical HCU caused by deficiency of the pyridoxine-dependent enzyme, cystathione beta-synthase, which converts homocysteine to cystathionine in the transsulphuration pathway. An alternative route for homocysteine metabolism is its remethylation to methionine by the cobalamin-dependent enzyme, methionine synthase, using the folate derivative, methyltetrahydrofolate, as a methyl donor. Remethylation defects are caused by impaired activity of methionine synthase itself, of an enzyme required to generate its methylcobalamin cofactor from dietary vitamin B12, or of the enzyme methyltetrahydrofolate reductase (MTHFR), which generates the methyl donor.
The correct diagnosis can be inferred from additional laboratory investigations including a complete blood count and quantitation of methionine and methylmalonic acid. Methionine is high/normal in HCU and low in the remethylation disorders. In the latter, cobalamin defects are readily distinguished from MTHFR by a coexisting macrocytic anaemia and further delineated by presence or absence of methylmalonic acid in urine or plasma.
Lowering homocysteine reverses thromboembolic risk. In HCU, this may be achieved with pyridoxine alone or with betaine as an alternative methyl donor. Some patients additionally follow a methionine-restricted diet. Betaine is the primary treatment for MTHFR and cobalamin disorders are managed with high-dose hydroxocobalamin.
- Folic Acid
- Genetic Diseases, Inborn
- Vitamin B 12
Statistics from Altmetric.com
Introduction
Homocystinuria (HCU) comprises a number of disorders of homocysteine (HCy) metabolism united by the common biochemical finding of a high blood HCy concentration. Hyperhomocystinaemia is implicated in the prothrombotic tendency and neurological features associated with all of these conditions.1 Therefore, lowering HCy is a major goal of treatment which in turn necessitates an understanding of the range of differential diagnoses of hyperhomocystinaemia to ensure the correct treatment is initiated.
Textbooks tend to consider the conditions under separate headings (often in separate chapters) as disorders of vitamin B12, folate and sulphated amino acid metabolism. While this is logical from a biochemical perspective, it is not always helpful for the clinician requesting measurement of HCy who, faced with a high result, must not necessarily conclude that their patient has ‘classical HCU’. In this article, we will guide the reader through the differential diagnosis of hyperhomocystinaemia and the management of each of the inherited disorders of HCy metabolism.
Biochemistry of HCy metabolism
HCy is generated from the essential amino acid, methionine in the transsulphuration pathway which thereafter utilises the vitamin B6 (pyridoxine)-dependent enzyme cystathione β-synthase (CBS) to convert HCy to cystathionine for the synthesis of cysteine. Remethylation of HCy to methionine via a vitamin B12 (cobalamin)-dependent pathway utilising the folate derivative, 5-methyltetrahydrofolate (5-MTHF), as a methyl donor (figure 1) presents an alternative route for HCy metabolism. The latter is essential for multiple processes including DNA methylation, and synthesis of creatine, choline and epinephrine (figure 1).

Homocysteine is generated from methionine in the transsulphuration pathway and metabolised to cysteine by pyridoxine (B6)-dependent cystathionine β-synthase (CBS). Remethylation of homocysteine to methionine is catalysed by the methylcobalamin (MeCbl) dependent enzyme, methionine synthase (MS) using the folate derivative, 5-methyltetrahydrofolate (5-MTHF) as a methyl donor. Methylcobalamin is generated from dietary cobalamin. Betaine is an alternative methyl donor utilising betaine-homocysteine methyltransferase (BHMT). Inherited disorders causing high homocysteine are shown in bold red adjacent to affected enzyme: HCU, classical homocystinuria; disorders of cobalamin metabolism, cbl F, cblJ, cblC, cblD, cblE, cblG. 5, 10-MeTHF, 5, 10-methyltetrahydrofolate; AdoCbl, adenosylcobalamin; AMN, amnionless; CUBN, cubilin and cobalamin lysosomal transport proteins; GIF, gastric intrinisic factor; MMACHC, methylmalonic aciduria homocystinuria type C protein; MT, Methyltransferase; MTHFR, methyltetrahydrofolate reductase; MUT, methylmalonylcoA mutase; SAH, S-adenosyl homocysteine; SAM, S-adenosyl methionine; TCII, transcobalamin-2.
Primary genetic disorders affecting function of CBS (classical HCU) or enzymes involved in remethylation impair metabolism of HCy causing it to rise in plasma from a reference concentration below 15 μmol/L to at least 40 μmol/L and usually over 100 μmol/L. A modestly elevated total HCy (tHCy) of 15–40 mmol/L is more likely attributable to a secondary (non-genetic) cause2 (table 1) relating to vitamin B12 or folate deficiency which may occur as part of a global malabsorption disorder (eg, coeliac disease and short bowel syndrome) or a specific deficit of either vitamin. Exposure to drugs or toxins causes a functional vitamin deficit in the context of a normal serum concentration, notably nitrous oxide which causes irreversible oxidation of the cobalt atom in vitamin B12 and use of the folate antagonist, methotrexate, without concomitant folate supplementation. Kidney failure causes a high HCy because of impaired renal excretion.
Differential diagnosis of a high plasma homocysteine
There is considerable overlap in tHCy concentration between primary and secondary causes of hyperhomocystinaemia. Therefore, while tHCy concentration can be used as a guide as to the potential underlying aetiology, it should be interpreted in the context of the clinical history and additional investigations, which will help to gauge the degree of clinical suspicion of a genetic disorder of HCy metabolism and exclude secondary causes of hyperhomocystinaemia.
Laboratory diagnosis
tHCy analysis is the quantitation of HCy in plasma following chemical reduction of thiol bonds using, for example, dithiothreitol. In normal plasma, the majority of HCy is protein bound and free HCy disulphide (fHCy) concentrations are negligible.3 fHCy becomes rapidly protein bound in vitro and accurate quantitation therefore requires plasma to be deproteinised within 30 min of venepuncture. Furthermore fHCy is only detectable once tHCy is greater than about 60 μmol/L4 making it a less sensitive marker than tHCy. These limitations of fHCy measurement and the widespread availability of tHCy assays have meant measurement of fHCy has now been largely superseded by tHCy. Whole blood is collected into lithium-heparin or EDTA. Analytical methods in routine use include HPLC, HPLC-tandem mass spectrometry and automated immunoassays. An artefactual rise in tHCy caused by its in vitro release from blood cells occurs at a rate of about 10% per hour at room temperature and about 10% over 24 hours at 4°C.5 Therefore, samples must ideally be transported to the laboratory on ice or otherwise separated within 1 hour of venepuncture. No special precautions around fasting or dietary protein intake are needed prior to sampling, though prandial status and recent protein intake may account for the small non-clinically significant diurnal fluctuations in tHCy concentration observed in studies.2
To aid interpretation of an abnormal tHCy result, samples for tHCy should be accompanied by contemporaneous samples for complete blood count (CBC) to include haemoglobin (Hb) and mean cell volume (MCV), and for vitamin B12, folate and renal function.
The most common causes of a high tHCy relate to underlying vitamin B12 or folate deficiency which is often first suspected when the CBC reveals a macrocytic anaemia.
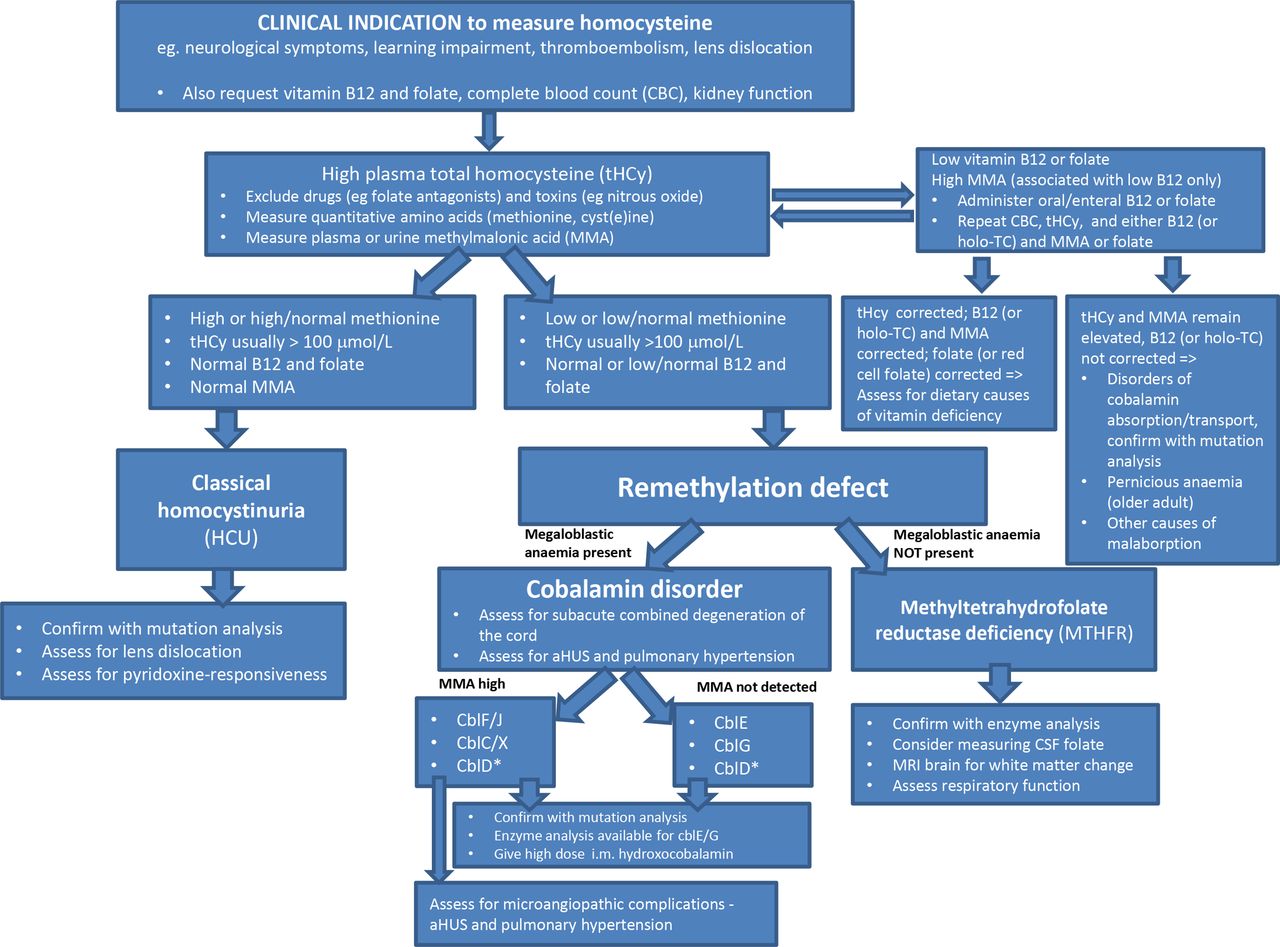
In a minority of cases serum B12 and folate results may not reflect whole body deficits and if deficiency is suspected, confirmatory tests are indicated. In suspected folate deficiency this is by measurement of red cell folate.6 Clinically significant B12 deficiency is confirmed by high urine or plasma methylmalonic acid (MMA) or measurement of holo-transcobalamin (holo-TC), which represents the active cobalamin fraction bound by TC.7 Correction of biochemical parameters following enteral vitamin B12 or folate administration differentiates problems of absorption and metabolism from nutritional deficiencies (figure 2).

{kind=link}
{kind=link}
Algorithm for further evaluation of a high plasma homocysteine. CblD* association with high MMA depends on the affected enzyme subunit. aHUS, atypical haemolytic uraemic syndrome; cbl, cobalamin defect; holo-TC, holotranscobalamin; i.m., intramuscular; MMA, methylmalonic acid.
Once secondary causes are excluded, measurement of other amino acids in the HCy metabolic pathway, notably methionine and cystine, with quantitation of MMA in plasma or urine helps to distinguish between the genetic conditions of classical HCU and remethylation defects caused by MTHF reductase (MTHFR) or a disorder of cobalamin metabolism. In HCU, remethylation is the only route available for HCy metabolism, and consequently, methionine is high and cystine, distal to the defective CBS enzyme, is low. Conversely, in remethylation disorders, CBS is the only route available for HCy metabolism, therefore, methionine is low. In the remethylation disorders caused by defects of cobalamin metabolism, there is an accompanying megaloblastic anaemia. The cobalamin disorders are further delineated by the presence or absence of MMA in plasma or urine (figure 2).
Until recently, a clinical and biochemical diagnosis would have been confirmed by measurement of fibroblast enzyme activity or complementation analysis. However, rapid advancements in genetics have meant that fibroblast studies have been largely superseded by mutation analysis. The limitations to next generation sequencing (NGS) strategies8 include incorrect gene panel selection, pathogenic intronic variants not detected by exome sequencing, insufficient sequencing depth and coverage, false negative results in presence of large deletions and interpretation of variants of uncertain significance. Thus, fibroblast studies retain utility if NGS has not been informative and clinical suspicion remains high.
Prospective or very early initiation of treatment of classical (non-pyridoxine-responsive) HCU before complications arise dramatically improves the outlook for patients and formed the rationale for its inclusion in newborn bloodspot screening programmes in many countries worldwide.9 Screening programmes employ raised methionine concentrations as the primary marker for HCU owing to the chemical reduction step for tHCy measurement being poorly amenable to high-throughput analysis. Bloodspot tHCy when used as a second tier test (as employed in the UK) enables the methionine threshold for a positive result to be lowered, increasing the screening sensitivity while reducing the false positive rate. Despite this, most pyridoxine-responsive forms of HCU will not be detected as methionine is not sufficiently elevated. Additionally, the UK newborn screening programme is not designed to identify remethylation disorders, though some countries screen for these using methionine concentrations below a defined lower limit.
Clinical conditions of HCU
HCy measurement is indicated as part of the routine evaluation of a child presenting with developmental delay or with neurological features including movement disorder, seizures and myelopathy. It is also indicated in the investigation of an individual at any age with lens dislocation or an unprovoked thromboembolic event.
Classical HCU
HCU caused by CBS deficiency is the most common condition of hyperhomocystinaemia. Its estimated worldwide prevalence from newborn screening and clinical case finding is between 1 in 200 000 and 335 000,10 11 although it varies by ethnicity with a higher prevalence seen in certain populations (1 in 65 000 in Ireland, 1 in 17 800 in Germany and 1 in 1800 in Qatar).
Thromboembolic tendency increases in direct proportion to the magnitude and duration of HCy elevation. Left untreated there’s a 30% chance of a vascular event by age 20 rising to 50% by the age of 30.12 Lens subluxation is a hallmark of HCU distinguishing it from remethylation disorders and is detectable in approximately 70% of untreated patients by the age of 10 and in over 90% diagnosed later.12 It causes a stepwise deterioration in visual acuity which may go unnoticed or be attributed to other causes unless specifically examined for. Additional ophthalmic complications of HCU include severe myopia, strabismus, glaucoma, retinal detachment and optic atrophy. Failing to consider a diagnosis of HCU in a patient presenting with lens subluxation or an unprovoked thromboembolic event leads to significant delays in initiating appropriate treatment to prevent further complications.13 14
Skeletal and connective tissue disorders affect approximately 50% of HCU patients, and may variably include elongated long bones (‘Marfanoid’ body habitus), arachnodactyly, joint laxity, osteopenia, scoliosis and vertebral collapse.
Learning disabilities are not universal or may be subtle and, when present, include global intellectual impairment, specific learning disabilities such as autism and behavioural disturbance.
HCU treatment is summarised in table 2. The primary goal is to maintain plasma tHCy concentration at less than 100 μmol/L in adults and less than 50 μmol/L in children at which thromboembolic risk is reversed, lens subluxation is halted and bone growth in children proceeds normally.15 16 Very early initiation of treatment allows for normal intellectual development, but once learning difficulties are evident they are not reversed by treatment.
Treatment and monitoring of the disorders of homocystinuria
The CBS enzyme uses pyridoxal 5’-phosphate as a cofactor. Approximately 50% of patients with HCU respond partially or wholly to high-dose pyridoxine (vitamin B6). Pyridoxine responsiveness is thought to depend on the extent to which the CBS mutations affect the cofactor binding site. Notably, the p.I278T missense mutation, present in approximately 25%, confers pyridoxine responsiveness even in the heterozygous state.17 People who are homozygous for this mutation and nutritionally replete may remain asymptomatic without treatment. Conversely, the p.G307S mutation prevalent in the Irish population is not associated with pyridoxine responsiveness.18 Testing for pyridoxine responsiveness forms an essential part of the diagnostic process for any patient newly diagnosed with HCU (figure 2). Briefly, this involves correcting dietary B12 or folate deficiency which may otherwise impair response to pyridoxine, ensuring adequate dietary protein intake, and administering high-dose pyridoxine (100–500 mg daily depending on age) with repeat measurement of tHCy after 1 week. A suggested protocol is detailed in the 2017 guideline.15
In fully pyridoxine-responsive HCU, target tHCy can be attained with oral pyridoxine alone (table 2) typically with doses in excess of the recommended daily allowance, ranging from 300 to 900 mg/day in adults and 10 mg/kg/day to a maximum of 500 mg/day in children.15 More commonly patients are partially responsive at high doses (postpyridoxine tHCy <80% baseline but >50 μmol/L). Pyridoxine excess is associated with peripheral neuropathy; therefore, it is ideally prescribed under specialist supervision with patients routinely monitored for this side effect. For the other 50% who do not respond to pyridoxine at all (postpyridoxine tHCy >80% baseline) pyridoxine does not form part of their treatment regimen.
If the tHCy target is not attainable with pyridoxine alone, oral betaine (N,N,N-trimethylglycine) is given as a methyl donor which provides an additional route for remethylation of HCy to methionine catalysed by betaine-homocysteine methyl transferase (figure 1). High methionine levels in patients on betaine can rarely lead to cerebral oedema presenting symptomatically with raised intracranial pressure or asymptomatically as white matter change on brain MRI.19 20 Therefore, the dose used is the maximum that allows for maintenance of methionine at less than 1000 μmol/L (which in adults may be higher than the UK licensed dose of 3 g twice daily). Patients who are partially pyridoxine-responsive can often be adequately and safely treated with a combination of pyridoxine and betaine only (table 2).
In non-pyridoxine-responsive HCU and some partially responsive patients, target tHCy can only be attained by additionally following a protein restricted diet to limit intake of the essential amino acid, methionine, the precursor to HCy production in the transsulphuration pathway (figure 1).12 21 Protein intake typically ranges from 5 to 40 g daily, with the shortfall met by taking a methionine-free amino acid supplement enriched with vitamins and minerals and additional cystine. The low protein diet is complex to manage and difficult to adhere to and patients need expert dietetic support throughout life without which they may not only fail to maintain tHCy at target but are also at risk of nutritional deficiencies.
In all forms of HCU, routine supplementation with folate and vitamin B12 is advisable as a low risk non-evidence-based intervention to prevent nutritional deficiencies of these vitamins contributing to treatment resistance. Their routine use may also enhance the remethylation pathway for HCy metabolism. Additional supportive measures include optician and ophthalmology input for eyesight and lens correction, bone density surveillance, and where necessary, lifelong educational and social support.
Remethylation defects
Remethylation defects are caused by MTHFR deficiency (known as 'MTHFR') and disorders of cobalamin metabolism. Neurological features are prominent often from an early age and include developmental delay, seizures, encephalopathy, dystonia, ataxia and hypotonia or hypertonia. There is considerable overlap between conditions which precludes making a reliable diagnosis on the basis of clinical presentation alone.22
However, attenuated forms are increasingly recognised. In MTHFR, there may be a period of normal development followed by regression of previously acquired intellectual and motor skills. Cerebral atrophy and white matter change is often evident on brain MRI. Sudden respiratory failure is described and contributes to a poor outcome in both early and later onset forms of untreated MTHFR.23 24
Cobalamin disorders may present later with progressive neurological features such as sensory deficit, weakness and gait disturbance caused by subacute combined degeneration of the cord as the clinical manifestation of functional vitamin B12 deficiency, or with thromboembolic complications associated with hyperhomocystinaemia. The cobalamin disorders associated with high MMA have a well-recognised late presentation with microangiopathic complications of atypical haemolytic uraemic syndrome (aHUS) and pulmonary artery hypertension.25
MTHFR deficiency
MTHFR enzyme converts 5,10-MTHF to 5-MTHF which is the methyl group donor for synthesis of methionine from HCy (figure 1). Hence in MTHFR, high tHCy is accompanied by a low/low-normal methionine (figure 2). The metabolic block does not affect the availability of folate for nucleic acid synthesis which accounts for why there is no associated megaloblastic anaemia.
Direct enzyme assays are available for confirmation of the diagnosis. Mutational analysis should be interpreted with caution as there are a number of MTHFR polymorphisms described, including the extensively studied thermolabile variant, c.677C>T which, with coexisting dietary folate deficiency, contribute to a modestly elevated tHCy (40–100 μmol/L, table 1). These variants may contribute to risk of neural tube defects in the developing fetus of affected mothers26 but do not cause the clinical features associated with true MTHFR.
In this condition, 5-MTHF, distal to the metabolic defect, is low. 5-MTHF is the only folate derivative available to the central nervous system and hence both 5-MTHF and methionine levels in cerebrospinal fluid (CSF) are low (figure 1). The methionine deficit, rather than hyperhomocystinaemia, is implicated in the prominent neurological features seen in MTHFR even at modest degrees of tHCy elevation compared with other disorders. Hence, in addition to lowering tHCy, a major goal of treatment is to increase availability of methionine to the brain (table 2). This is primarily achieved with betaine which does not itself cross the blood–brain barrier but rather, by acting as an alternative methyl donor, increases the peripheral supply of methionine which does enter CSF. There is a substantial evidence base demonstrating its benefit in both improved survival and neurological development.27 Betaine also reduces plasma tHCy and the associated risk of vascular complications.
High-dose folate or its derivatives also form part of the treatment regimen but there remains no clear consensus as to the optimal formulation. The metabolic defect means that 5-MTHF cannot be generated from folate or folinic acid and therefore biochemically it is logical to administer 5-MTHF directly as calcium mefolinate which then enters the CSF.28 However, in vivo instability of mefolinate means that, even at high doses, it does not completely correct CSF 5-MTHF concentration. No neurological outcome studies demonstrating clinical efficacy of mefolinate vs other forms of folate in MTHFR have been conducted. However, in other disorders of folate metabolism associated with low CSF 5-MTHF, small increases, not amounting to complete correction, have been shown to improve neurological outcomes.29
Nitrous oxide administered as an anaesthetic agent has been reported to have catastrophic consequences in undiagnosed MTHFR30 31 and its use is contraindicated in this condition.
Disorders of cobalamin metabolism
Vitamin B12 is required to generate the methylcobalamin (MeCbl) co-factor for methionine synthase which catalyses the conversion of HCy to methionine. Hence, cobalamin disorders directly or indirectly impair methionine synthase activity resulting in high tHCy accompanied by a low or low/normal methionine (figure 1). They are accompanied by a macrocytic anaemia, with or without neutropenia or pancytopaenia, because accumulated 5-MTHF traps folate and disrupts bone marrow nucleotide synthesis. They are grouped into disorders of absorption and transport, in which serum B12 is low and urine/plasma MMA is elevated and disorders of intracellular cobalamin metabolism in which there is a functional B12 deficit with normal serum vitamin B12. In the latter, presence of MMA in plasma or urine depends on the defect. Diagnostic confirmation of the genetic disorders is by mutation analysis, though enzyme analysis is available for two of the intracellular cobalamin disorders (cbl E and G).
Inherited disorders of cobalamin absorption and transport include mutations in gastric intrinsic factor, the cubam receptor subunits cubilin and amnionless (Imerslund-Gräsbeck syndrome) and TC-2 (figure 1).32–35 They typically present in childhood with neurological manifestations of B12 deficiency. Pernicious anaemia caused by autoantibodies to intrinsic factor required for vitamin B12 absorption is the most common acquired disorder and presents later in life.
Recreational nitrous oxide misuse is increasingly common and its presentation can mimic that of an intracellular cobalamin disorder with subacute neurological features of functional B12 deficiency (although on a background of normal neurological development), high tHCy and MMA and normal serum vitamin B12 concentration.36
The inherited intracellular disorders of cobalamin metabolism are denoted cblC-G, cblJ and cblX in brackets after the affected enzyme. LMBD1 (cblF) and ABCD4 (cblJ) are transporter proteins, which release stored cobalamin from hepatocyte lysosomes. Thereafter cobalamin is processed by methylmalonic aciduria homocystinuria type C protein (MMACHC) (cblC) which is dependent on a transcription regulatory complex (cblX) and directed by the enzyme, MMADHC (cblD), either towards synthesis of MeCbl by methionine synthase reductase (cblE) or towards synthesis of adenosylcobalamin (AdoCbl)37–43 (figure 1).
Plasma and urine MMA is concurrently high in cblC, some forms of cblD depending on the affected MMADHC subunit, and the cobalamin disorders upstream of MMACHC (cblX, F, J). This arises from dual impairment of AdoCbl synthesis, the cofactor for the enzyme, methylmalonyl CoA mutase, required for methylmalonyl coA metabolism. In these conditions, dual elevation of MMA and tHCy appears to exert a ‘multitoxic’ effect that causes microangiopathic complications of aHUS and pulmonary hypertension which is not observed in the other cobalamin disorders (cbl E, G and some forms of cblD) in which MMA is not elevated.22 Cobalamin disorders which only impact on AdoCbl synthesis cause high MMA and normal HCy. CblC is the most common inherited cobalamin disorder, with an estimated prevalence of 1 in 100 000. The others are much rarer with only a small number of cases described.
The outcome in all cobalamin disorders is dramatically improved by prompt initiation of high-dose parenteral hydroxocobalamin, initially 1 mg/day intramuscularly (table 2) and later titrated down to the minimum dose required to achieve a target tHCy level and to normalise Hb and MCV.44 Betaine may be added to improve biochemical tHCy and methionine response. Methionine supplementation is considered if a normal range methionine level is not achieved with hydroxocobalamin and betaine. There are several reports of microangiopathic complications associated with cblC being halted or even reversed by treatment25 45 and an isolated case report of a patient with cblC in whom prospective treatment of the mother with hydroxocobalamin during pregnancy and the baby from birth prevented onset of neurological complications.46 The authors are aware of a similarly favourable outcome in a prospectively treated patient with cblG following the diagnosis in an older sibling.
General practitioners, if asked to prescribe hydroxocobalamin, must be made aware of the very high doses used to treat cobalamin disorders and cautioned against measuring vitamin B12 levels to monitor treatment adherence or to guide the prescribing dose.
Summary
There is considerable overlap in the clinical manifestation of the disorders of HCy metabolism discussed in this article all of which may present with neurological and cognitive symptoms of variable severity and/or with unprovoked thromboembolism. Lens subluxation is notable as a unique feature of HCU while microangiopathic features are only seen in a subset of cobalamin disorders associated with high MMA. The widespread availability of the tHCy assay has contributed to greater awareness of HCU as a potential cause for such presentations but secondary causes of hyperhomocystinaemia relating to vitamin B12 or folate deficiency must also be considered, and clinicians may be not be familiar with the remethylation disorders which equally form part of the differential diagnosis. Making the correct diagnosis is dependent on judicious requesting and interpretation of additional laboratory tests to ensure that correct and timely treatment is initiated which dramatically improves clinical outcome.
Ethics statements
Patient consent for publication
Acknowledgments
We are grateful to Dr Subadra Wanninayake for preparing figure 1.
References
Footnotes
Handling editor Patrick J Twomey.
Contributors Both authors have contributed equally to the writing of this article.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- Viewpoint