
Abstract
The arrival of targeted therapies has presented both a conceptual and a practical challenge in the treatment of patients with advanced non-small cell lung carcinomas (NSCLCs). The relationship of these treatments with specific histologies and predictive biomarkers has made the handling of biopsies the key factor for success. In this study, we highlight the balance between precise histological diagnosis and the practice of conducting multiple predictive assays simultaneously. This can only be achieved where there is a commitment to multidisciplinary working by the tumor board to ensure that a sensible protocol is applied. This proposal for prioritizing samples includes both recent technological advances and the some of the latest discoveries in the molecular classification of NSCLCs.
Similar content being viewed by others

Introduction
The classification of primary lung carcinomas is probably one of the least precise among all the solid tumors. Paradoxically, however, it is one of the most reproducible. The lung pathology field has not only been static, as recently mentioned, but also perceived as uninteresting [1]. This has come about due to a set of factors which can be better understood from a historical perspective. The discovery that some of these carcinomas, i.e., small cell carcinomas (SCLCs), responded incredibly well to chemotherapy led to a lack of interest in the correct classification of lung carcinomas. It is a situation that continues even today. This therapeutic discovery gave rise to the use of the intelligent term “non-small cell lung carcinoma (NSCLC)” and to a tolerance of the lack of precision. According to Edwards et al., this nomenclature was suggested in 1984 by Chuang et al. and accepted from then on during the pre-immunohistochemistry (IHC) era to decrease the risk of over-interpreting small biopsy specimens [2–4]. Over the years, the term has been extended to surgical specimens, clinical trials, etc. [5, 6].
The term “large cell carcinoma (LCC)” was defined by the 2004 WHO classification as one of exclusion [7]. It is also misused for two main reasons:
-
(a)
In the setting of surgical specimens, it gives rise to the possibility of giving such a name to all difficult cases with no obvious keratin (squamous cell carcinomas, SCCs) or gland formation (adenocarcinomas, ACs).
-
(b)
It is frequently applied as a synonym of non-small cell carcinoma in small thoracic biopsies [7–9].
A few years ago, there was a sudden interest, which has continued to grow, in the histology of lung cancer [10]. This can be explained by various factors. Firstly, ACs have treatable molecular alterations: mainly EGFR mutations and ALK translocation [11–14]. Secondly, some targeted agents should not be used in SCCs. This is not only because they do not provide better response rates (pemetrexed), but also because their use in this histological type is associated with life-threatening complications (i.e., bevacizumab) [15–17]. As if we had not learned our lesson previously, another imprecise term is emerging: “non-squamous histology” [15, 18].
As such, the current therapeutic situation of lung cancer demonstrates that establishing a treatment requires a precise histological diagnosis. Many treatable molecular alterations are linked to specific histological types. Equally, diagnosis must be reasonable. If we exhaust the sample during classification, we will not be able to produce predictive biomarkers [19, 20]. Certain considerations are, therefore, necessary. Although there appears to be complete specificity for AC or SCC for several driver mutations, some poorly differentiated ACs may be classified as SCC due to the presence of squamoid features [21]. Moreover, a significant number of those poorly differentiated ACs may have treatable molecular alterations [22]. For example, up to 18 % of so-called squamoid subtype ACs can harbor EGFR mutations [23]. The practical implication is that it is probably better to use the “NSCLC-NOS” (not otherwise specified) category when there is inconclusive evidence of squamous or glandular differentiation.
Lung carcinoma classification for non-pathologists
In the paragraphs that follow, we shall discuss certain aspects which are, perhaps, less well-known to clinicians or pathologists with less experience in thoracic pathology.
While SCLCs are not the subject of this study, it is useful to include a paragraph about them. Currently, we recommend confirmation with IHC of all SCLCs which appear as such in the study with hematoxylin and eosin (H&E), i.e., scant cytoplasm, granular nuclear chromatin with nucleoli inconspicuous or absent and high mitotic rate. For the IHC confirmation, the following are sufficient: two neuroendocrine differentiation markers (CD56, chromogranin or synaptophysin); a pankeratin AE1–AE3 with frequent “dot-like” pattern; TTF-1 (70–90 % positive) and a very high proliferative index by Ki-67 (70–100 %). This is very important in making the differential diagnosis with lymphoma, melanoma, and other lung neoplasias: poorly differentiated SCCs, large neuroendocrine cell carcinoma, and carcinoid tumors, especially [24, 25]. Two aspects must be taken into account:
-
(a)
Neoplasias with neuroendocrine differentiation can show positivity for p63. This includes some 77 % of SCLCs [26].
-
(b)
SCLCs observed outwit the typical bronchial biopsy with crush artifact, and chromatin stretching have an appearance with which we, pathologists, are not familiar, i.e., very well-conserved cells with visible nucleus, greater size, etc. Examples might include SCLCs observed in a pulmonary core-needle biopsy, in a wedge lung resection specimen or in a metastasis. These unfamiliar appearances can present huge difficulties in differential diagnosis [25].
Furthermore, ACs and SCCs represent the two major types of NSCLCs. Distinguishing between the two histological types can appear extremely difficult by routine light microscopy, particularly in small biopsies and cytology samples. This can affect up to 35 % of cases [27]. In the case of NSCLCs, the use of IHC is essential where a specific, conclusive diagnosis cannot be produced. Indeed, we can only make such a diagnosis where we identify keratinization in SCCs or gland formation in ACs. As such, probably the most accepted antibody pair in the literature is that formed by TTF-1 (marker of glandular differentiation) and p63 (marker of squamous differentiation) [28]. In additional, SCCs also tend to be positive for desmocollin-3 (the most specific marker) and CK5/6 (the most sensitive marker) [27]. ACs, however, stain with Napsin A (the most specific marker) and CK7 (the most sensitive marker) [27]. Interestingly, desmocollin-3 (DCS3) was the top differentially expressed gene in our own microarray comparison between lung ACs and SCCs [29]. This finding has been independently confirmed by other groups [27, 30].
It must be stressed that analytical and post-analytical aspects (interpretation) can influence the supposed specificity and sensitivity of an antibody. Given this consideration, it is useful, for example, to recall that genuine staining of p63 must be intensive and extensive. Faint or focal immunostaining for p63 should be considered non-specific until there is proof that it is not [9, 27, 28, 31–35]. Recent studies show the value of p40 (∆Np63) as a more specific marker of squamous differentiation [36–38]. Another idea is to use double staining protocols combining a nuclear and a cytoplasmic/membranous antibody (for example, CK7-p63 and CK5/6-TTF-1) to reduce the number of slides necessary [27]. However, we should always bear in mind that methodologies taken to raise specificity may also lower the likelihood of clinical application. For example, new antibodies or protocols may not be easy to implement or interpret.
Sample prioritization
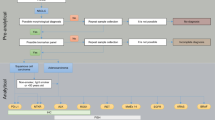
While we firmly believe as pathologists in the importance of the correct histological subtyping of carcinomas of the lung, we also understand that it is not always possible in the real world. There is currently no consensus on how to prioritize the different predictive assays that are frequently performed in different laboratories using different approaches. These include, for example, H&E and IHC for diagnosis, accurate histological subtyping and some predictive biomarkers, fluorescence in situ hybridization (FISH) for ALK translocation, and PCR for EGFR mutations [35, 39, 40]. The “tissue sparing” algorithm that we propose for molecular tests in small thoracic samples is depicted in Fig. 1 [41–43]. This approach not only saves time and tissue, but will also provide realistic information on the true incidence and overlap of the different molecular alterations. These considerations are not merely academic. Rather, they influence the cost of drugs and their biomarkers [44].

A realistic approach for sample prioritization for the study of predictive biomarkers in patients with advanced lung ACs. Route A is for cases that require classificatory IHC while route B is for cases that are diagnosed based on the H&E alone. The relative frequency of the different genetic alterations is shown in parenthesis. Data from ROS1 translocation is taken from the literature [43]. The other percentages come from our own experience [41, 42], and unpublished data
In order to minimize the loss of material, the following are suggested:
-
1.
Sufficient multidisciplinary communication, oncological, and pathological to put the paraffin block in the microtome as few times as possible;
-
2.
Reasonable use of classificatory IHC with a restricted panel of antibodies. While this increases the percentage of “NSCLC-NOS”, the sample is preserved for future studies on therapeutic targets [27, 35].
Figure 2 shows how sample prioritization and biomarker information are integrated into patient care.

A simplified view of an integrated pathway for the study of predictive biomarkers in patients with advanced NSCLCs. There are two interesting aspects. a If the sampling is managed by the tumor board, there will be better sample prioritization than has happened previously. b There is an increased awareness of technological advantages and disadvantages among patients and their families
Predictive biomarkers
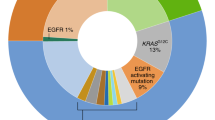
As a result of our experience in recent years, we believe that a protocol similar to that depicted in Fig. 1 would provide information on multiple predictive biomarkers [22, 28, 41, 42, 45]. Although some aspects of our approach have already been implemented, others remain theoretical [42]. Ideally, the protocol should be validated prospectively with an intention-to-treat philosophy. Our protocol has several advantages. Firstly, the sample is sectioned in as few steps as possible to decrease tissue waste at the microtome and also to shorten the turn-around time. Secondly, this approach permits the combination of different methodologies: IHC, in situ hybridization and PCR. This is important because, currently, it is not realistic to “multiplex” the analytical phase of, for example, mutation, amplification, translocation, and overexpression detection. Thirdly, it allows the order of the different biomarkers to be changed depending on estimated prevalence, histological characteristics, drug approvals or access to clinical trials, or compassionate use. For example, ACs with lepidic (formerly “bronchioloalveolar”), papillary or micropapillary components are commonly associated with EGFR mutations, papillary growth has also been linked with BRAF V600E mutated ACs, and signet-ring cells are typically present in ALK positive lung ACs [46–49]. Fourthly, it also allows the methodology to be changed. This is feasible because most IHC and in situ hybridization tests are performed on 3–5 μm sections, and there are recent PCR methods that allow mutation testing from a single 5 μm section [50–52]. Fifthly, simultaneous testing of at least some of the biomarkers (those included in step A3/B2 and step A4/B3) will give us the response as to whether they are truly mutually exclusive or not, as well as the true prevalence in a given population. If they are not mutually exclusive, we will not leave potentially positive patients without testing. This is especially important now as, where methodological caveats can be reasonably discounted, we are beginning to accept the existence of:
-
(a)
Molecular heterogeneity: recently published discordance rates between primary tumors and metastases were 14 % for EGFR mutations and 13.5 % for ALK overexpression [53, 54];
-
(b)
Overlap in predictive markers. For example, ALK translocations may be identified in both EGFR and HER2 mutant patients [55, 56].
Ideally, an H&E is conducted first to confirm the malignant nature of the disease. If the tumor shows malignant glands, an AC can be diagnosed with certainty. If not, a couple of classificatory IHC stains is probably sensible (step A2). Step A3/B2 involves simultaneous testing for EGFR mutations and for ALK translocation. Almost everyone will agree that, in patients with advanced lung ACs, it is necessary to know those two biomarkers. However, there is no consensus on whether testing should be simultaneous or sequential. Nor is there consensus on the methodology to be used [35, 39, 57, 58]. A wide variety of methods have been applied to EGFR mutation analysis. Although direct sequencing is still probably the most frequently used method, in recent years commercial real time PCR assays have become increasingly popular [41, 45]. Although FISH is currently the only approved method to identify potential responders to crizotinib through the presence of the ALK translocation, ALK IHC may also prove useful in this setting [57].
The remaining biomarkers (step A4/B3) represent a more innovative approach to lung cancer targeted therapies. It is probably wise to check the status of KRAS [59]. In addition, patients with ROS rearrangements may also respond to crizotinib [43]. We should also extract DNA again and look for mutations in HER2 and BRAF [60–62]. A final H&E section will re-assure us as to how many tumor cells we have left for the latter assays and the possibility of producing more biomarkers in the future (i.e., MET amplification, MET overexpression, and RET rearrangement) [63–65].
To conclude, the final consideration is: what is the minimum number of tumor cells needed to conduct these tests? For IHC, counts of 2,000 tumor cells are recommended [66]. In the case of FISH, 100 cells are sufficient [50, 66]. For PCR techniques, it is very important to know their limit of detection (LOD) and to work with a safety margin. For example, if the LOD of direct sequencing is 30 %, the percentage of tumor cells should ideally be 40–50 % [41, 45, 66, 67]. In the case of real time PCR with a LOD of 1–5 %, we must be aware of the risk of false negatives when the percentage of tumor cells is below 10 % [41, 45, 66, 67].
Unfortunately, most of the advances in personalized treatment of NSCLCs have been confined to the treatment of patients with ACs. However, better molecular characterization of SCCs is enabling this subgroup to become a growing area of interest. As such, specific molecular defects such as FGFR1 amplifications, DDR2 mutations, PI3KCA amplifications and mutations will become part of the routine molecular diagnostic workup of SCCs [68–71]. Lessons learned from ACs should help to make these exciting findings a success story for the treatment of SCCs.
In conclusion, we have presented a realistic approach to lung AC targeted therapies. This includes accurate histological subtyping, as well as sample prioritization to release information on as many predictive biomarkers as possible.
References
Travis WD, IASLC Staging Committee (2009) Reporting lung cancer pathology specimens. Impact of the anticipated 7th Edition TNM classification based on recommendations of the IASLC Staging Committee. Histopathology 54:3–11
Edwards SL, Roberts C, McKean ME et al (2000) Preoperative histological classification of primary lung cancer: accuracy of diagnosis and use of the non small cell category. J Clin Pathol 53:537–540
Chuang MT, Marchevsky A, Teirstein AS et al (1984) Diagnosis of lung cancer by fibreoptic bronchoscopy: problems in the histological classification of non-small cell carcinomas. Thorax 39:175–178
Thomas JS, Lamb D, Ashcroft T et al (1993) How reliable is the diagnosis of lung cancer using small biopsy specimens? Report of a UKCCCR Lung Cancer Working Party. Thorax 48:1135–1139
Abratt RP, Lee JS, Han JY et al (2006) Phase II trial of gemcitabine-carboplatin-paclitaxel as neoadjuvant chemotherapy for operable non-small cell lung cancer. J Thorac Oncol 1:135–140
Pfister DG, Johnson DH, Azzoli CG, American Society of Clinical Oncology et al (2004) American Society of Clinical Oncology treatment of unresectable non-small-cell lung cancer guideline: update 2003. J Clin Oncol 22:330–353
Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC (2004) Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. World Health Organization (WHO) Classification of Tumours. IARC Press, Lyon
Travis WD, Brambilla E, Noguchi M et al (2011) International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 6:244–285
Kerr KM (2012) Personalized medicine for lung cancer: new challenges for pathology. Histopathology 60:531–546
Langer CJ, Besse B, Gualberto A et al (2010) The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol 28:5311–5320
Lynch TJ, Bell DW, Sordella R et al (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of nonsmall-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139
Paez JG, Jänne PA, Lee JC et al (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500
Pao W, Miller V, Zakowski M et al (2003) EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 101:13306–13311
Kwak EL, Bang YJ, Camidge DR et al (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363:1693–1703
Scagliotti GV, Parikh P, von Pawel J et al (2008) Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol 26:3543–3551
De Marinis F, Pereira JR, Fossella F et al (2008) Lung Cancer Symptom Scale outcomes in relation to standard efficacy measures: an analysis of the phase III study of pemetrexed versus docetaxel in advanced non-small cell lung cancer. J Thorac Oncol 3:30–36
Sandler A, Gray R, Perry MC et al (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small cell lung cancer. N Engl J Med 355:2542–2550
Kubota K, Niho S, Enatsu S et al (2009) Efficacy differences of pemetrexed by histology in pretreated patients with stage IIIB/IV non-small cell lung cancer: review of results from an open-label randomized phase II study. J Thorac Oncol 4:1530–1536
Hirsch FR, Wynes MW, Gandara DR et al (2010) The tissue is the issue: personalized medicine for non-small cell lung cancer. Clin Cancer Res 16:4909–4911
Dacic S (2011) Lung carcinoma morphology or mutational profile: that is the question. Arch Pathol Lab Med 135:1242–1243
Rekhtman N, Paik PK, Arcila ME et al (2012) Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clin Cancer Res 18:1167–1176
Conde E, Angulo B, Tang M et al (2006) Molecular context of the epidermal growth factor receptor mutations: evidence for the activation of mTOR/S6 K signaling. Clin Cancer Res 12:710–717
Wilkerson MD, Yin X, Walter V et al (2012) Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomalinstability, and methylation. PLoS One 7:e36530
Travis WD (2012) Update on small cell carcinoma and its differentiation from squamous cell carcinoma and other non-small cell carcinomas. Mod Pathol 1:S18–S30
Leslie KO, Wick MR (2011) Practical pulmonary pathology. A diagnostic approach. Churchill Livingstone, Philadelphia
Au NH, Gown AM, Cheang M et al (2004) P63 expression in lung carcinoma: a tissue microarray study of 408 cases. Appl Immunohistochem Mol Morphol 12:240–247
Warth A, Muley T, Herpel E et al (2012) Large-scale comparative analyses of immunomarkers for diagnostic subtyping of non-small-cell lung cancer biopsies. Histopathology 61:1017–1025
Conde E, Angulo B, Redondo P et al (2010) The use of P63 immunohistochemistry for the identification of squamous cell carcinoma of the lung. PLoS One 5:e12209
Angulo B, Suarez-Gauthier A, Lopez-Rios F et al (2008) Expression signatures in lung cancer reveal a profile for EGFR-mutant tumours and identify selective PIK3CA overexpression by gene amplification. J Pathol 214:347–356
Monica V, Ceppi P, Righi L et al (2009) Desmocollin-3: a new marker of squamous differentiation in undifferentiated large-cell carcinoma of the lung. Mod Pathol 22:709–717
Mukhopadhyay S, Katzenstein AL (2011) Subclassification of non-small cell lung carcinomas lacking morphologic differentiation on biopsy specimens: utility of an immunohistochemical panel containing TTF-1, napsin A, p63, and CK5/6. Am J Surg Pathol 35:15–25
Pelosi G, Rossi G, Bianchi F et al (2011) Immunhistochemistry by means of widely agreed-upon markers (cytokeratins 5/6 and 7, p63, thyroid transcription factor-1, and vimentin) on small biopsies of non-small cell lung cancer effectively parallels the corresponding profiling and eventual diagnoses on surgical specimens. J Thorac Oncol 6:1039–1049
Terry J, Leung S, Laskin J et al (2010) Optimal immunohistochemical markers for distinguishing lung adenocarcinomas from squamous cell carcinomas in small tumor samples. Am J Surg Pathol 34:1805–1811
Sterlacci W, Savic S, Schmid T et al (2012) Tissue-sparing application of the newly proposed IASLC/ATS/ERS classification of adenocarcinoma of the lung shows practical diagnostic and prognostic impact. Am J Clin Pathol 137:946–956
Thunnissen E, Kerr KM, Herth FJ et al (2012) The challenge of NSCLC diagnosis and predictive analysis on small samples. Practical approach of a working group. Lung Cancer 76:1–18
Bishop JA, Teruya-Feldstein J, Westra WH et al (2012) p40 (ΔNp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma. Mod Pathol 25:405–415
Pelosi G, Fabbri A, Bianchi F et al (2012) ΔNp63 (p40) and thyroid transcription factor-1 immunoreactivity on small biopsies or cellblocks for typing non-small cell lung cancer: a novel two-hit, sparing-material approach. J Thorac Oncol 7:281–290
Nonaka D (2012) A study of ΔNp63 expression in lung non-small cell carcinomas. Am J Surg Pathol 36:895–899
Mino-Kenudson M, Mark EJ (2011) Reflex testing for epidermal growth factor receptor mutation and anaplastic lymphoma kinase fluorescence in situ hybridization in non-small cell lung cancer. Arch Pathol Lab Med 135:655–664
Garrido P, de Castro J, Concha A et al (2012) Guidelines for biomarker testing in advanced non-small-cell lung cancer. A national consensus of the Spanish Society of Medical Oncology (SEOM) and the Spanish Society of Pathology (SEAP). Clin Transl Oncol 14:338–349
Angulo B, Conde E, Suárez-Gauthier A et al (2012) A comparison of EGFR mutation testing methods in lung carcinoma: direct sequencing real-time PCR and immunohistochemistry. PLoS One 7:e43842
Conde E, Angulo B, Izquierdo E et al (2012) The ALK translocation in advanced NSCLCs: pre-approval testing experience at a Single Cancer Center. Histopathology. doi:10.1111/his.12037
Bergethon K, Shaw AT, Ou SH et al (2012) ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 3:863–870
Atherly AJ, Camidge DR (2012) The cost-effectiveness of screening lung cancer patients for targeted drug sensitivity markers. Br J Cancer 106:1100–1106
Angulo B, Conde E, Martínez R et al (2011) Analytic performance of a novel real-time PCR method for the detection of mutations in EGFR in formalin-fixed paraffin-embedded tissue (FFPET) samples of lung cancer. J Thorac Oncol 6:S1061
Yatabe Y, Kosaka T, Takahashi T et al (2005) EGFR mutation is specific for terminal respiratory unit type adenocarcinoma. Am J Surg Pathol 29:633–639
Motoi N, Szoke J, Riely GJ et al (2008) Lung adenocarcinoma: modification of the 2004 WHO mixed subtype to include the major histologic subtype suggests correlations between papillary and micropapillary adenocarcinoma subtypes, EGFR mutations and gene expressionanalysis. Am J Surg Pathol 32:810–827
Yousem SA, Nikiforova M, Nikiforov Y (2008) The histopathology of BRAF-V600E-mutated lung adenocarcinoma. Am J Surg Pathol 32:1317–1321
Yoshida A, Tsuta K, Nakamura H et al (2011) Comprehensive histologic analysis of ALK-rearranged lung carcinomas. Am J Surg Pathol 35:1226–1234
Thunnissen E, Bubendorf L, Dietel M et al (2012) EML4-ALK testing in non-small cell carcinomas of the lung: a review with recommendations. Virchows Arch 461:245–257
Cobas DNA Sample Preparation Kit CE-IVD package insert 2011
Cobas EGFR Mutation Kit CE-IVD package insert 2011
Chen ZY, Zhong WZ, Zhang XC et al (2012) EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist 17:978–985
Kim H, Xu X, Yoo S-B et al (2013) Discordance between anaplastic lymphoma kinase status in primary non-small cell lung cancer and their corresponding metastases. Histopathology 62:305–314
Kelly RJ, Carter CA, Giaccone G (2012) HER2 Mutations in Non-Small-Cell Lung Cancer Can Be Continually Targeted. J Clin Oncol 30:3318–3319
Doebele RC, Pilling AB, Aisner DL et al (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 18:1472–1482
Wallander ML, Geiersbach KB, Tripp SR et al (2012) Comparison of reverse transcription-polymerase chain reaction, immunohistochemistry, and fluorescence in situ hybridization methodologies for detection of echinoderm microtubule-associated protein like 4-anaplastic lymphoma kinase fusion-positive non-small cell lung carcinoma: implications for optimal clinical testing. Arch Pathol Lab Med 136:796–803
Pao W, Ladanyi M (2007) Epidermal growth factor receptor mutation testing in lung cancer: searching for the ideal method. Clin Cancer Res 13:4954–4955
Oxnard GR, Jänne PA (2012) KRAS wild-type lung cancer: a moving target in an era of genotype migration. J Clin Oncol 30:3322–3324
Arcila ME, Chaft JE, Nafa K et al (2012) Prevalence, clinicopathologic associations and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res 18:4910–4918
Paik PK, Arcila ME, Fara M et al (2011) Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 29:2046–2051
Gautschi O, Pauli C, Strobel K et al (2012) A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. J Thorac Oncol 7:e23–e24
Ou SH, Kwak EL, Siwak-Tapp C et al (2011) Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol 6:942–946
Spigel DR, Ervin TJ, Ramlau R et al (2010) Randomized multicenter double blind placebo controlled phase II study evaluating MetMAb, an antibody to Met receptor, in combination with erlotinib, in patients with advanced non-small-cell lung cancer. Ann Oncol 21 (abstr LBA5146)
Takeuchi K, Soda M, Togashi Y et al (2012) RET, ROS1 and ALK fusions in lung cancer. Nat Med 18:378–381
Eberhard DA, Giaccone G, Johnson BE et al (2008) Biomarkers of response to epidermal growth factor receptor inhibitors in Non-Small-Cell Lung Cancer Working Group: standardization for use in the clinical trial setting. J Clin Oncol 26:983–999
Pirker R, Herth FJ, Kerr KM et al (2010) Consensus for EGFR mutation testing in non-small cell lung cancer: results from a European workshop. J Thorac Oncol 5:1706–1713
Gold KA, Wistuba II, Kim ES (2012) New strategies in squamous cell carcinoma of the lung: identification of tumor drivers to personalize therapy. Clin Cancer Res 18:3002–3007
Weiss J, Sos ML, Seidel D et al (2010) Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2:62ra93
Hammerman PS, Sos ML, Ramos AH et al (2011) Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov 1:78–89
Janku F, Tsimberidou AM, Garrido-Laguna I et al (2011) PIK3CA mutations in patients with advanced cancers treated with PI3 K/AKT/mTOR axis inhibitor. Mol Cancer Ther 10:558–565
Acknowledgments
This work was partially funded by Fundación Mutua Madrileña and Fondo de Investigaciones Sanitarias (PI11/02866). Translated into English by Michelle Homden.
Conflict of interest
None declared.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Conde, E., Angulo, B., Izquierdo, E. et al. Lung adenocarcinoma in the era of targeted therapies: histological classification, sample prioritization, and predictive biomarkers. Clin Transl Oncol 15, 503–508 (2013). https://doi.org/10.1007/s12094-012-0983-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-012-0983-z