
Abstract
Discrepancies have been reported between HER2 status in primary breast cancer and micrometastatic cells in bone marrow. The aim of this study was to assess HER2 gene status in micrometastatic cells in bone marrow and corresponding primary tumour. Micrometastatic cells were detected in bone marrow aspirations in a prospective series of 27 breast cancer patients by immunocytochemistry (pancytokeratin antibody). HER2 status of micrometastatic cells was assessed by fluorescence in situ hybridisation (FISH), respectively in 24 out of 27. Primary tumour HER2 status was assessed by immunohistochemistry (CB11 antibody) and by FISH in 20 out of 27 of the cases. HER2 was amplified or overexpressed in five out of 27 (18.5%) primary tumours and in four out of 27 (15%) micrometastatic cells. In two cases, HER2 was overexpressed and amplified in primary tumour, but not in micrometastatic cells, whereas, in one case, HER2 presented a low amplification rate (six copies) in micrometastatic cells not found in the primary tumour. We demonstrated that negative and positive HER2 status remained, in the majority of the cases, stable between the bone marrow micrometastasis and the primary tumour. Therefore, the efficiency of anti-HER2 adjuvant therapy could be evaluated, in a clinical trial, by sequential detection of HER2-positive micrometastatic cells within the bone marrow, before and after treatment.
Similar content being viewed by others

Main
HER2 overexpression, observed in 15–30% of breast cancers, is associated with a poor outcome, especially in node-positive breast carcinoma (Gusterson et al, 1992; Perou et al, 2000). HER2 status remains stable between the primary tumour site and distant metastasis (Niehans et al, 1993; Gancberg et al, 2002; Vincent-Salomon et al, 2002; Carlsson et al, 2004) or regional lymph node metastasis (Simon et al, 2001). In contrast, HER2 has been found to be overexpressed in 60–100% in bone marrow micrometastatic cells, independently of the primary tumour status (Braun et al, 2001b). Cytotoxic agents currently used for chemotherapy in high-risk breast cancer patients do not completely eliminate micrometastatic cells in bone marrow (Braun et al, 2000a) and bone marrow micrometastasis in breast cancer patients is associated with a poor outcome (Braun et al, 2000b, 2005; Naume et al, 2004). In this context, a targeted therapy, specific for micrometastatic cells, would be appropriate. However a recent study showed HER2 heterogeneous overexpression in bone marrow micrometastatic cells could be detected in patients with HER2-negative primary tumours. This heterogeneity may reduce the efficacy of an immunotherapy-based strategy in an adjuvant setting (Solomayer et al, 2006). The aim of this pilot study was to assess HER2 gene status by fluorescence in situ hybridisation (FISH) in micrometastatic cells in bone marrow of breast cancer patients and to compare it to HER2 primary tumour status, in order to evaluate if anti-HER2 therapy in adjuvant setting, could be given to patients after an assessment of HER2 status of the primary tumour only. In addition, as the cytokeratin antigens detected in epithelial micrometastatic cells are not specific to cancer cells, morphological analysis of positive detected cells is a major step in the identification of micrometastatic cells. In this perspective, the second aim of our study was to confirm that the isolated cytokeratin-positive (CK+) cells detected in bone marrow aspirates and interpreted as micrometastasis actually corresponded to tumour cells. We therefore documented the neoplastic nature of the cells by assessing the gene status of other frequently amplified oncogenes in breast carcinomas (Taccagni et al, 1997; Al-Kuraya et al, 2004; Orsetti et al, 2004), especially CCND1 (cyclinD1) and MYC.
Materials and methods
Patients
Cytokeratin positive micrometastatic cells were detected in bone marrow aspirates in a prospective series of breast cancer patients. Bone marrow samples positive for micrometastatic cells and the corresponding primary tumours were obtained from 27 patients (three stage II, one stage III, one local relapse and 22 stage IV). A single bone marrow aspiration was performed under local anaesthesia from the posterior iliac crest before chemotherapy in an adjuvant setting or for metastatic disease. Informed consent was obtained from all patients.
Bone marrow specimens
Techniques have been described previously (Pierga et al, 2004). Briefly, 3–5 ml of bone marrow aspirate was collected on EDTA (Vacutainer, Becton Dickinson, Le Pont de Claix, France). Components of the bone marrow aspirate from the two iliac crests were processed under sterile conditions. Each sample was diluted by addition of half the volume of Hanks solution (Gibco BRL, Invitrogen, Cergy, Pontoise, France). Samples were separated by Ficoll/Hypaque density centrifugation (Sigma, St Louis, MO, USA; density, 1.077 g ml−1) in Leucosep tubes (Polylabo, Au Verney, Servion, France) (830 g, 15 min, 20°C). The mononuclear cells (MNCs) layer was harvested from each tube, combined, diluted in 50 ml of Hanks and centrifuged at 360 g, 5 min at 20°C. Cells were resuspended in PBS/0.1% bovine serum albumin (BSA). After dilution to 3% in pure acetic acid for red cell lysis, an aliquot of the cell suspension was counted. The MNCs were resuspended in PBS/BSA at 1.106 ml−1. One millilitre of the cell suspension was cytocentrifuged twice onto polylysine-coated slides at 580 g for 3 min (Hettich Universal 16A cytocentrifuge). The supernatant was carefully removed from each slide after the first cytocentrifugation and the slides were allowed to dry in air overnight. Slides were stored at −20°C and then at −80°C until staining.
Immunocytochemical staining
The pancytokeratin (CK) monoclonal antibody A45-B/B3 (Micromet, Munich, Germany and Chromavision, San Juan, Capistrano, USA), which recognises several cytokeratin epitopes which characterise CK 8, CK 18 and CK 19, was applied for cell detection (Stigbrand et al, 1998). The immunostaining procedure was standarised by using an automated device (Cadenza, Shandon, France). Before staining, cytospots were fixed with 4% paraformaldehyde for 5 min, and then dried for 15 min at room temperature. Endogenous alkaline phosphatase was then blocked with TBS solution (Sigma) with 2% human AB serum, for 15 min. This solution was used to dilute primary and secondary antibodies. After blocking, the slides were incubated with the primary antibody A45 B/B3 (2 μg ml−1 for 40 min). Control slides were incubated under the same conditions with a mouse monoclonal anti-FITC IgG1 (1/1250) (Sigma). Slides were incubated for 20 min with secondary polyclonal rabbit anti-mouse antibody (Dako France, Trappes, France, A/S, Glostrup, Denmark). After each step, the slides were rinsed for 5 min in TBS 1 X solution. Immune complexes were revealed by the alkaline phosphatase-anti-alkaline phosphatase (APAAP) technique (Dako) (1/50) for 25 min. The chromogen reaction was performed for 20 min with a colorimetric substrate of fuchsin solution (2.5% in 2 N HCl) (New Fuchsin, Sigma) with 4% NaNO2, 8% β-naphthol (Sigma) and 2% levamisole (Dako). Cells were counterstained with Mayer hematoxylin (1 min) (Sigma) diluted to one out of three in distilled water. The specimen was then rinsed under running water for 5 min and then in TBS. Slides were coverslipped using Faramount mounting medium (Dako). Mononuclear cells (3 × 106) in three slides were evaluated for each patient. Negative controls, stained with anti-FITC monoclonal mouse antibody, were performed on an equivalent number of cells (i.e. three slides, 3 × 106 mononuclear cells) for each patient.
Positive controls were obtained with bone marrow from ‘normal’ donors undergoing orthopedic surgery, spiked with SKBR3 or MCF7 cell lines, 10–102 for 106 mononuclear cells per cytospot. One positive control slide and one negative control slide were added to each series of 20 stained slides in the automated device.
CK+ cell detection
Cell detection was performed by manual screening with an optical microscope. Criteria for evaluation of immunostained cells in bone marrow were adapted from Borgen et al (1999) based on the results of the European ISHAGE Working Group for standarisation of tumour cell detection. The main criteria were a large cell size, a high nuclei/cytoplasm ratio and the absence of obvious haematopoietic cell morphology.
HER2 status
HER2 status of micrometastatic cells in BM was assessed by FISH on slides on which CK+ cells had been detected. Slides were rinsed in PBS, then treated by pepsin (0.05% in 0.01 N HCl), for 5 min at 37°C, then dehydrated in ethanol series. Digoxigenin-labelled HER2 probe solution (Zymed Laboratories Inc., South San Francisco, CA, USA) was laid onto the slides, which were covered by coverslips. In some cases, directly SpectrumOrange-labelled Cyclin D1 or myc probes (Vysis, Downers Grove, IL, USA) were added to the mix in order to increase the probability of detecting abnormal micrometastatic cells. Simultaneous denaturation of the probes and cell DNA was performed at 75°C for 2 min. Slides were then incubated overnight at 37°C in humid chamber, for hybridisation. Rinsing was performed in 0.4 × SSC/0.3% Igepal at 75°C, for 4 min, then in the same solution at 20°C, for 2 min, followed by 5 min in PBS. HER2 hybrids were revealed by incubation with a FITC-labeled anti-digoxigenin antibody (Roche Diagnostics, Basel, Switzerland), 1/100 dilution, for 30 min, at 37°C. Finally, slides were mounted in Vectashield/DAPI (Vector Laboratories, Burlingame, CA, USA). Preparations were analysed by microscope and when technically possible, all CK+ cells detected by their cytoplasmic fluorescence were photographed under FITC, and, in the case of Cyclin D1 or myc hybridisation, under SpectrumOrange excitations.
HER2 status of primary tumour was assessed by immunohistochemistry (CB11 antibody, Novocastra, Newcastle, UK) and by FISH in 20 cases out of the 27 cases, with available and suitable blocks for HER2 FISH analysis.
HER2 immunostainings and FISH were performed on histological tissue sections prepared from a representative sample of the primary tumour. Immunohistochemical procedures for the analysis of HER2 expression were defined to provide a strong correlation between HER2 overexpression and gene amplification status, as determined by FISH (Couturier et al, 2000). After rehydration and antigenic retrieval in citrate buffer (10 mM, pH 6.1), tissue sections were incubated with the CB11 anti-p185 HER/neu monoclonal antibody (Novocastra, Newcastle UK), for 1 h, at 1/800 dilution. Staining was revealed with the Vectastain Elite ABC peroxidase mouse IgG kit (Vector Burlingame, CA, USA), using diaminobenzidine (Dako A/S, Glostrup, Denmark) as chromogen. Under these conditions, normal epithelial cells were not immunostained and therefore constituted an internal negative control.
Immunostainings were scored as strong, weak or negative according to the percentage of labelled tumour cells and membrane staining intensity. Cases were considered to be positive when at least 60% of cells were stained (Bilous et al, 2003; Vincent-Salomon et al, 2003). HER2 status was then classified as overexpressed (strong or moderate staining) or not significantly overexpressed.
Fluorescence in situ hybridisation was performed according to the same protocol as that already described earlier for micrometastasis. In addition, after deparaffinisation, slides were first treated with a protein digesting enzyme, at 37°C, for 10 min.
Cytokeratins 8/18 expression was also assessed on these 20 out of 27 primary tumours according to a previously published protocol (Azoulay et al, 2005).
Results
Patient characteristics are summarised in Table 1. The mean age was 55.6 years (range: 36–75 years). Tumours were invasive ductal carcinomas in 85% of cases with a histological grade II or III in 14/25 (56%).
A visceral metastasis was observed 22 of the cases (22/27 cases, 81%). Nineteen cases presented bone metastases (19/27, 70% of cases). Eight of 22 stage IV patients had synchronous metastasis at primary diagnosis. Patients with metastatic disease received chemotherapy as first-line treatment in 65% of cases (15 patients), or second-line treatment (four patients)(17.5%) or third-line treatment (four patients) (17.5%).
HER2 status in primary tumours and bone marrow micrometastasis are summarised in Table 2. 6/27 (22.2%) primary tumours had HER2 overexpression (2+ and 3+). In five cases, the intensity of staining was strong (3+) and observed in 100% of tumour cells of the invasive component. In 4/4 (100%) cases assessable for FISH analysis, this overexpression was associated to HER2 amplification. The remaining case with moderate (++) staining did not show any amplification by FISH. Therefore, 5/27 (18.5%) tumours presented HER2 amplification and overexpression.
Micrometastatic cells in bone marrow were observed in all selected cases. The cell morphology on cytospots was interpreted according to the ISHAGE criteria for tumour cells. The cells were large, with a high nucleus/cytoplasm ratio. In 19 out of 27 cases, the micrometastatic cells formed clusters. The number of micrometastatic cells ranged from 1 to 1500 per slide examined.
In four out of 27 (15%) (95%CI: 2–28%) cases HER2 amplification was observed in bone marrow micrometastatic cells. When more than 20 cells were observed and interpretable the amplification was homogeneous. In two cases, HER2 was amplified and overexpressed in the primary tumour, but not in distant BM micrometastatic cells and in one case, HER2 presented a very low level of amplification in BM micrometastatic cells (six copies) and not in the primary tumour (two copies). At least, only two (50%) of four cases with HER-2 amplification in primary tumours showed HER-2 amplification in micrometastatic cells, and three (75%) of four cases with HER-2 amplification in micrometastatic cells showed HER-2 amplification in the primary tumours.
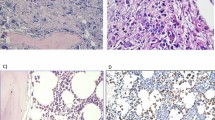
CCND1 gene status was assessed in 20/27 of the cases. CCND1 was amplified in seven of these 20 cases (35%). Notably, four of these seven CCND1 amplified cases showed also an amplification of HER2 (Figure 1). In the remaining three cases, this amplification confirmed that the detected HER2-negative CK+ cells actually corresponded to tumour cells.

One micrometastatic cell CK+ (intracytoplasmic red labelling) with HER 2 amplification (red spots) and CCND1 amplification (green spots).
MYC gene status was assessed in six cases and was amplified in one case, in which HER2 and CCND1 were also amplified (Table 2).
Four cases, without HER2 amplification, presented three HER2 gene copies per nucleus and thus demonstrated a HER2 overrepresentation in relation with a chromosome 17 trisomy. The presence of chromosome 17 trisomy was another proof of malignancy of these micrometastatic cells (Figure 2) (Table 2).

Chromosome 17 trisomy (blue spots) in CK+ cells (cytoplasmic red labelling) and disomy (blue spots) in cytokeratine-negative cell.
All analysed cases showed cytokeratin 8/18 expression in their primary breast tumour, ranging from 10 to 100% of positive cells per case.
Discussion
In this pilot study, we wanted to document the stability of the HER2 status between primary tumours and their bone marrow micrometastasis. We observed that the majority of the HER2-negative tumours were associated with HER2-negative micrometastasis except in one case in which micrometastatic cells demonstrated a lower level of HER2 amplification. In addition, in our series of breast carcinomas, 15% of bone marrow micrometastatic cells presented HER2 amplification. This rate is very close to that observed in primary breast tumours. HER2 gene amplification appears thus to occur before bone marrow micrometastatic process in breast cancer and to remain stable during bone marrow micrometastatic spread.
This result is in accordance with those concerning visceral metastases and local and regional metastases (Barnes et al, 1988; Niehans et al, 1993; Masood and Bui, 2000; Simon et al, 2001; Gancberg et al, 2002; Vincent-Salomon et al, 2002; Carlsson et al, 2004). Recently, in a meta-analysis of the published data concerning HER2 status stability among primaries and metastases, Carlsson et al (2004) confirmed that there was no drastic modification in HER2 status between primary tumours and their locoregional lymph node metastases and their distant visceral metastases.
Our results and these published data on the stability of HER2 status between primary and metastatic tumours are in contrast with the recently published studies by Schmidt-Kittler et al (2003) and by Klein et al (Mercapide et al 2002) showing that micrometastatic cells demonstrated fewer chromosomal alterations, such as losses and gains detected by single-cell CHG analysis, than primary tumour cells (Mercapide et al 2002; Schmidt-Kittler et al, 2003). Another recently published study also showed that HER2 amplification was more frequently observed in circulating cells than in primary tumours and therefore concluded that HER2 amplification could be acquired during the metastatic process (Meng et al, 2004). The HER2 amplicon corresponds to a 280-kb minimal region of amplification at the HER2 locus of chromosome 17q arm, in breast cancer. The amplification is significantly associated with increased expression of six of the 10 genes located within this region (HER2, GRB7, PNMT, MLN64, MGC9753 and MGC 1483) as described by Kauraniemi et al (2003). This amplicon has been observed since the early stage of in situ carcinoma (Van de Vijver et al, 1988). The observation of HER2 amplification exclusively in disseminated cells, suggests a selection of clones within the primary tumour that harboured initial HER2 amplification and that were underrepresented in the primary tumour, for example, by cytotoxic agents, rather than an acquisition of this amplification de novo within the metastatic cells.
Previous studies by Pantel et al (1993, Braun et al (2001a) and Solomayer et al (2006), reported high rates of HER2 overexpression ranging from 60 to 100% of cases, analysed by immunocytochemistry only. These rates could therefore be explained by technical aspects, that is, excessively sensitive immunohistochemistry technique. Using the FISH approach, we demonstrated that primary breast tumours and bone marrow micrometastasis demonstrate the same range of HER2 activation, ranging around 15% of the cases (Table 3).
The gene expression profile of metastasis and their primaries has also been compared and has been shown to cluster together (Ramaswamy et al, 2003; Weigelt et al, 2003). In the literature, Cyclin D1 amplification in primary breast tumours ranged between 10 and 23%. In our series, the CyclinD1 (CCND1) amplification rate was 35% (Al-Kuraya et al, 2004). This rate is therefore higher than previously reported for ductal carcinomas, although our series is very small to derive any conclusions about this issue. However, we can speculate that the amplification rate might be higher in this group of metastatic and advanced breast carcinomas. Amplification of HER2 with other oncogenes has been reported previously, particularly in a recently published FISH study (Al-Kuraya et al, 2004). In this work, HER2 was associated with CCND1 amplification in 18% of cases. CCND1 was amplified in 20% of cases. Determination of coamplification rates of major oncogenes such as MYC and CCND1 in breast carcinomas should provide important information regarding prognosis. It has also been recently reported, during the 2005 San Antonio meeting, that MYC status could be a predictive parameter of tumour response to anti-HER2 therapy (Gianni et al, 2005).
In conclusion, the HER2 status assessed by FISH in isolated micrometastatic cancer cells in bone marrow was well correlated with that of primary tumour. Our pilot study showed that in the majority of the cases, the stable, positive or negative, status of HER2 during the bone marrow micrometastatic process. This observation on a small series of cases should be confirmed on a larger scale and identification of HER2-positive micrometastatic cells in breast carcinomas could constitute part of patient management in a future clinical trial, in order to select patients for anti-HER2 adjuvant therapy. Repeated assessment of the presence of micrometastases could also be part of the follow-up and evaluation of the efficacy of anti-HER2 therapy.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Al-Kuraya K, Schraml P, Torhorst J, Tapia C, Zaharieva B, Novotny H, Spichtin H, Maurer R, Mirlacher M, Kochli O, Zuber M, Dieterich H, Mross F, Wilber K, Simon R, Sauter G (2004) Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res 64: 8534–8540
Azoulay S, Lae M, Freneaux P, Merle S, Al Ghuzlan A, Chnecker C, Rosty C, Klijanienko J, Sigal-Zafrani B, Salmon R, Fourquet A, Sastre-Garau X, Vincent-Salomon A (2005) KIT is highly expressed in adenoid cystic carcinoma of the breast, a basal-like carcinoma associated with a favorable outcome. Mod Pathol 18: 1623–1631
Barnes DM, Lammie GA, Millis RR, Gullick WL, Allen DS, Atman DG (1988) An immunohistochemical evaluation of c-erbB-2 expression in human breast carcinoma. Br J Cancer 58: 448–452
Bilous M, Dowsett M, Hanna W, Isola J, Lebeau A, Moreno A, Penault-Llorca F, Ruschoff J, Tomasic G, van de Vijver M (2003) Current perspectives on HER2 testing: a review of national testing guidelines. Modern Pathol 16: 173–182
Borgen E, Naume B, Nesland JM, Kvalheim G, Beiske K, Fodstad O, Diel I, Solomayer EF, Theocharous P, Coombes RCS, BM, Wunder E, Marolleau J-P, Garcia J, Pantel K (1999) Standardization of the immunocytochemical detection of cancer cells in BM and blood: I. establishment of objective criteria for the evaluation of immunostained cells. Cytotherapy 1: 377–388
Braun S, Cevatli BS, Assemi C, Janni W, Kentenich CRM, Schindlbeck C, Rjosk D, Hepp F (2001a) Comparative analysis of micrometastasis to the bone marrow and lymph nodes of node-negative breast cancer patients receiving no adjuvant therapy. J Clin Oncol 19: 1468–1475
Braun S, Kentenich C, Janni W, Hepp F, de Waal J, Willgeroth F, Sommer H, Pantel K (2000a) Lack of effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high-risk breast cancer patients. J Clin Oncol 18: 80–86
Braun S, Pantel K, Muller P, Janni W, Hepp F, Kentenich CRM, Gastroph S, Wischnik A, Dimpfl T, Kindermann G, Riethmuller G, Schlimok G (2000b) Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N Engl J Med 342: 525–533
Braun S, Schlimok G, Heumos I, Schaller G, Riethdorf L, Riethmuller G, Pantel K (2001b) ErbB2 overexpression on occult metastatic cells in bone marrow predicts poor clinical outcome of stage I-III breast cancer patients. Cancer Res 61: 1890–1895
Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, Schlimok G, Diel IJ, Gerber B, Gebauer G, Pierga JY, Marth C, Oruzio D, Wiedswang G, Solomayer EF, Kundt G, Strobl B, Fehm T, Wong GY, Bliss J, Vincent-Salomon A, Pantel K (2005) A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med 353: 793–802
Carlsson J, Nordgren H, Sjostrom J, Wester K, Villman K, Bengtsson NO, Ostenstad B, Lundqvist H, Blomqvist C (2004) HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br J Cancer 90: 2344–2348
Couturier J, Vincent-Salomon A, Nicolas A, Zafrani B, Sastre-Garau X (2000) Strong correlation between results of fluorescent in situ hybridization and immunohistochemistry for the assessment of ERBB2 (HER-2/neu) gene status in breast carcinoma. Modern Pathol 13: 1238–1243
Gancberg D, Di Leo A, Cardoso F, Rouas G, Pedrocchi M, Paesmans M, Verhest A, Bernard-Marty C, Piccart MJ, Larsimont D (2002) Comparison of HER-2 status between primary breast cancer and corresponding distant metastatic sites 10.1093/annonc/mdf252. Ann Oncol 13: 1036–1043
Gianni L, Zambetti M, Clark K, Baker J, Cronin M, Wu J, Mariani G, Rodriguez J, Carcangiu M, Watson D, Valagussa P, Rouzier R, Symmans W, Fraser R, Jeffrey S, Hortobagyi GN, Pusztai L, Shak S (2005) Gene expression profiles in paraffin-embedded core biopsy tissue predict response to chemotherapy in women with locally advanced breast cancer. J Clin Oncol 23: 7265–7277
Gusterson BA, Gelber RD, Goldhirsch A, Price KN, Save-Soderborgh J, Anbazhagan R, Styles J, Rudenstam CM, Golouh R, Reed R, Martinez-Tello F, Tiltman A, Grigolato P, Bettelehim R, Neville AM, Bürki K, Castiglione M, Collins J, Lindtner J, Stenn H-J (1992) Prognostic importance of c-erbB-2 expression in breast cancer. International (Ludwig) Breast Cancer Study Group. J Clin Oncol 10: 1049–1056
Kauraniemi P, Kuukasjarvi T, Sauter G, Kallioniemi A (2003) Amplification of a 280-kilobase core region at the ERBB2 locus leads to activation of two hypothetical proteins in breast cancer. Am J Pathol 163: 1979–1984
Masood S, Bui MM (2000) Assessment of Her2/neu overexpression in primary breast cancers and their metastatic lesions: an immunohistochemical study. Annals Clin Laboratory Sci 30: 259–265
Meng S, Tripathy D, Shete S, Ashfaq R, Haley B, Perkins S, Beitsch P, Khan A, Euhus D, Osborne C, Frenkel E, Hoover S, Leitch M, Clifford E, Vitetta E, Morrison L, Herlyn D, Terstappen LW, Fleming T, Fehm T, Tucker T, Lane N, Wang J, Uhr J (2004) HER-2 gene amplification can be acquired as breast cancer progresses. Proc Natl Acad Sci USA 101: 9393–9398
Mercapide J, Zhang S, Fan X, Furio-Bacete V, Schneider J, Lopez de la Osa I, Patchefsky A, Klein-Szanto A, Castresana J (2002) CCND1- and ERBB2-gene deregulation and PTEN mutation analyses in invasive lobular carcinoma of the breast. Mol Carcinogen 35: 6–12
Naume B, Wiedswang G, Borgen E, Kvalheim G, Karesen R, Qvist H, Janbu J, Harbitz T, Nesland JM (2004) The prognostic value of isolated tumor cells in bone marrow in breast cancer patients: evaluation of morphological categories and the number of clinically significant cells. Clin Cancer Res 10: 3091–3097
Niehans GA, Singleton TP, Dykoski D, Kiang DT (1993) Stability of HER-2/neu expression over time and at multiple metastatic sites. J Natl Cancer Inst 85: 1230–1235
Orsetti B, Nugoli M, Cervera N, Lasorsa L, Chuchana P, Ursule L, Nguyen C, Redon R, du Manoir S, Rodriguez C, Theillet C (2004) Genomic and expression profiling of chromosome 17 in breast cancer reveals complex patterns of alterations and novel candidate genes. Cancer Res 64: 6453–6460
Pantel K, Schlimok G, Braun S, Kutter D, Lindemann F, Schaller G, Funke I, Izbicki JR, Riethmuller G (1993) Differential expression of proliferation-associated molecules in individual micrometastatic carcinoma cells. J Natl Cancer Inst 85: 1419–1424
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D (2000) Molecular portraits of human breast tumours. Nature 406: 747–752
Pierga JY, Bonneton C, Vincent-Salomon A, de Cremoux P, Nos C, Blin N, Pouillart P, Thiery JP, Magdelenat H (2004) Clinical significance of immunocytochemical detection of tumor cells using digital microscopy in peripheral blood and bone marrow of breast cancer patients. Clin Cancer Res 10: 1392–1400
Ramaswamy S, Ross KN, Lander ES, Golub TR (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33: 49–54
Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJF, Kaufmann M, Diebold J, Arnholdt H, Muller P, Bischoff J, Harich D, Schlimok G, Riethmuller G, Eils R, Klein CA (2003) From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA 100: 7737–7742
Simon R, Nocito A, Hübscher T, Bucher C, Torhorst J, Schraml P, Bubendorf L, Mihatsch M, Moch H, Wilber K, Schötzau A, Kononen J, Sauter G (2001) Patterns of HER-2/neu amplification and overexpression in primary and metastatic breast cancer. J Natl Cancer Inst 93: 1141–1146
Solomayer EF, Becker S, Pergola-Becker G, Bachmann R, Kramer B, Vogel U, Neubauer H, Wallwiener D, Huober J, Fehm TN (2006) Comparison of HER2 status between primary tumor and disseminated tumor cells in primary breast cancer patients. Breast Cancer Res Treat 98: 179–184
Stigbrand T, Andres C, Bellanger L, Bishr Omary M, Bodenmuller H, Bonfrer H, Brundell J, Einarsson R, Erlandsson A, Johansson A, Leca JF, Levi M, Meier T, Nap M, Nustad K, Seguin P, Sjodin A, Sundstrom B, van Dalen A, Wiebelhaus E, Wiklund B, Arlestig L, Hilgers J (1998) Epitope specificity of 30 monoclonal antibodies against cytokeratin antigens: the ISOBM TD5-1 workshop. Tumour Biol 19: 132–152
Taccagni G, Rovere E, Masullo M, Christensen L, Eyden B (1997) Myofibrosarcoma of the breast. Review of the literature on myofibroblastic tumors and criteria for defining myofibroblastic differentiation. Am J Surg Pathology 21: 489–496
Van de Vijver MJ, Peterse JL, Mooi WJ, Wisman P, Lomans J, Dalesio O, Nusse R (1988) Neu-protein overexpression in breast cancer. Association with comedo-type ductal carcinoma in situ and limited prognostic value in stage II breast cancer. N Engl J Med 319: 1239–1245
Vincent-Salomon A, Jouve M, Genin P, Fréneaux P, Sigal-Zafrani B, Caly M, Beuzeboc P, Pouillart P, Sastre-Garau X (2002) HER2 status in patients with breast carcinomas is not modified selectively by preoperative chemotherapy and is stable during the metastatic process. Cancer 94: 2169–2173
Vincent-Salomon A, Mac Grogan G, Couturier J, Arnould L, Mathoulin-Pélissier S (2003) Re: HER2 testing in the real world. J Natl Cancer Inst 95: 628–629
Weigelt B, Glas AM, Wessels LF, Witteveen AT, Peterse JL, van't Veer LJ (2003) Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci USA 100: 15901–15905
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Vincent-Salomon, A., Pierga, JY., Couturier, J. et al. HER2 status of bone marrow micrometastasis and their corresponding primary tumours in a pilot study of 27 cases: a possible tool for anti-HER2 therapy management?. Br J Cancer 96, 654–659 (2007). https://doi.org/10.1038/sj.bjc.6603584
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603584
Keywords
This article is cited by
-
EHD2 is a Predictive Biomarker of Chemotherapy Efficacy in Triple Negative Breast Carcinoma
Scientific Reports (2020)
-
Automated detection of the HER2 gene amplification status in Fluorescence in situ hybridization images for the diagnostics of cancer tissues
Scientific Reports (2019)
-
p63/MT1-MMP axis is required for in situ to invasive transition in basal-like breast cancer
Oncogene (2016)
-
Disseminated tumor cells as selection marker and monitoring tool for secondary adjuvant treatment in early breast cancer. Descriptive results from an intervention study
BMC Cancer (2012)
-
Breast cancer: is there a need for biopsy of metastases?
memo - Magazine of European Medical Oncology (2012)
