
Abstract
The complexity of the p53 protein, coupled with the vast cellular responses to p53, is simply astonishing. As new isoforms, functional domains and protein–protein interactions are described; each morsel of information forces us to think (and re-think) about how it ‘fits’ into the current p53 paradigm. One aspect of p53 signaling that is under refinement is the mechanism(s) leading to apoptosis. Here we discuss what is known about p53-induced apoptosis, what proteins and protein–protein interactions are responsible for regulating apoptosis, how can this cascade be genetically dissected, and what pharmacological tools are available to modulate p53-dependent apoptosis. While everything may not comfortably fit into our understanding of p53, all of these data will certainly broaden our viewpoint on the complexity and significance of the p53-induced apoptotic pathway. Here, our discussion is primarily focused on the works presented at the 12th International p53 Workshop, except where appropriate background is required.
Similar content being viewed by others

Basic Principles of Apoptosis
Apoptosis is a genetically controlled event with roles in tissue development,1 homeostasis and disease.2 It is defined by a pattern of molecular and morphological changes that result in the packaging and removal of the dying cell. Cells committed to die via apoptosis following developmental cues, stress or infection are removed by phagocytes to prevent a host immune response. Central to our understanding of the mechanism of apoptosis is the induction of caspase activity. Caspases are a family of cysteine proteases that orchestrate the dismantling and clearance of the apoptotic cell. Since the discovery that the Caenorhabditis elegans cell-death abnormality-3 (ced-3) gene product, which is required for apoptosis in nematodes, is homologous to the cysteine protease ICE (interleukin-1 converting enzyme), it has been recognized that caspases are required for the initiation and execution of apoptosis. There are two general signaling pathways that trigger apoptosis, the differences between these pathways dictate how the death signal is transduced, and thus, how the caspases become activated (Figure 1).

A comparison of the intrinsic and extrinsic apoptosis cascades. The intrinsic pathway is engaged by cellular stresses, such as DNA damage or growth factor withdrawal, and directly impacts on the Bcl-2 family of pro- and anti-apoptotic proteins. Members of this protein family can be transcriptionally-induced, transcriptionally-repressed or subjected to post-translational modifications that act to repress or enhance function. Once the appropriate repertoire of Bcl-2 proteins have been engaged, they elicit mitochondrial outer membrane permeabilization (MOMP), cytochrome c release and APAF-1-dependent pro-caspase-9 activation. Executioner caspases-3 and -7 are then activated by caspase-9-dependent cleavage. The extrinsic pathway requires the ligation of death receptors by death ligands, which results in the assembly of adaptor molecules and pro-caspase-8 activation. Again, executioner caspases-3 and -7 are then activated by caspase-8. Bid can also be cleaved (and activated) by caspase-8 leading to co-engagement of the intrinsic pathway
The intrinsic pathway (engaged by stresses such as DNA damage or hypoxia) is engaged by the transcriptional or post-translational regulation of Bcl-2 proteins that directly impact on mitochondrial outer-membrane permeabilization (MOMP).3 MOMP occurs through the action of pro-apoptotic multi-domain members such as Bcl-2-associated X protein (Bax) and Bcl-2 antagonist/killer (Bak); these proteins oligomerize through direct or indirect activation by the Bcl-2-homology domain-3 (BH3)-only, pro-apoptotic, Bcl-2 family members. After MOMP happens, cytochrome c is released from the mitochondrial intermembrane space, which causes APAF1 (apoptotic protease activating factor 1) oligomerization resulting in apoptosome formation. This complex, in turn, recruits and activates procaspase-9, which then activates executioner caspases-3 and -7. These caspases are responsible for the apoptotic hallmarks, such as chromatin condensation, plasma membrane asymmetry and cellular blebbing.
Death-receptor ligation by death ligands is responsible for the induction of the extrinsic pathway.4 For example, tumor necrosis factor (TNF) binds to its death receptor, TNFR1, which causes the recruitment of adaptor molecules, TRADD (TNFR1-associated death domain) and FADD (FAS-associated death domain), and the binding and activation of procaspase-8 molecules that are brought together by TRADD and FADD. Once caspase-8 is active, it can activate executioner caspases. Some cells do not die in response to the extrinsic pathway alone and require an amplification step that is induced by caspase-8. In this situation, another target of caspase-8 is the BH3-only protein, Bid (BH3-interacting-domain death agonist). Caspase-8 cleaved Bid (tBid-truncated) is then able to directly activate pro-apoptotic multi-domain proteins to induce MOMP, so this co-engages the intrinsic pathway.
Unifying p53 and Apoptosis: What Do We Know?
Cells that are insulted by oncogene expression, DNA damage or other forms of stress stabilize the p53 protein by phosphorylation or other modifications.5, 6 Stabilized p53 accumulates in the nucleus to regulate the expression of numerous pro-apoptotic genes (e.g., BAX, NOXA, PUMA, BID, CD95, APAF-1, DR5, p53AIP1).7, 8, 9, 10, 11, 12, 13, 14 Each of these genes when silenced or removed from a particular model system produced partial resistance to p53-induced apoptosis. Most likely, these genes govern the decision to live or die based on the cell type investigated and the applied death stimulus (meaning, each gene contributes to certain death pathways, but not all). While p53 functions as a transcription factor in the nucleus (there is certainly no doubt to this ability) it also possesses an extranuclear function to directly bind anti-apoptotic Bcl-2 proteins (Bcl-2 and Bcl-xL) and activate pro-apoptotic multi-domain Bcl-2 proteins (Bax and Bak) to regulate MOMP.15 Years of debate over the importance of the extranuclear function have led several laboratories to develop elegant systems to demonstrate this ‘unbelievable’ function of p53, and the amount of literature based on this aspect of p53 function is rapidly growing.15
While we do not understand each step of p53-dependent apoptosis, we certainly know how p53-dependent apoptosis presents itself. Cells that have engaged p53-dependent apoptosis typically follow the intrinsic cell death pathway (Figure 2). This pathway is regulated by pro-apoptotic Bcl-2 proteins, as Bcl-2 or Bcl-xL overexpression blocks this form of cell death. Mitochondria are the downstream target of pro-apoptotic Bcl-2 proteins, and these proteins function to permeabilize mitochondria. The pro-apoptotic factors that are released from mitochondria then activate caspases that produce the hallmarks of apoptosis (e.g., DNA laddering, asymmetry of the plasma membrane). Since caspases are responsible for dismantling the insulted cell, p53-dependent apoptosis can be postponed by the addition of caspase inhibitors; but because mitochondria are permeabilized and ATP production soon wanes, death is inevitable.

How p53 engages the apoptotic signal. Following cellular stress, p53 is subjected to numerous post-translational modifications that result in p53's stabilization and accumulation in the nucleus and cytoplasm. In the nucleus, p53 directly regulates the expression of numerous pro-apoptotic molecules, which are necessary for steps either directly before MOMP (e.g., BAX) or may potentially directly induce MOMP (e.g., PUMA, NOXA, BID). In the cytoplasm, p53 associates with Bcl-2 or Bcl-xL, which can release sequestered BH3-only proteins, or this interaction can inhibit p53's ability to directly activate pro-apoptotic Bcl-2 multi-domain proteins (i.e., Bax and Bak). Once MOMP occurs, apoptosome formation is initiated and the cell death cascade is engaged
Current studies focus on linking the functions of p53 to the apoptotic cascade by integrating all of p53's pro-apoptotic abilities, along with determining which proteins are responsible for generating those signals. While we don't have every piece of the cascade determined, our understanding of the p53-dependent mechanism(s) of apoptosis is becoming clearer.
Murine Models of p53-Induced Apoptosis: Defining the Elusive?
The numerous effects of p53 stabilization on maintaining tissue homeostasis and preventing aberrant cells from becoming tumors highlights the importance of understanding which functions (or combination of functions) of p53 are essential to its tumor suppressor activity (see review by Johnson and Attardi in this issue for a detailed discussion of this topic). The earliest animal models that examined p53 function in vivo focused on genetically disrupting the Trp53 gene, and these models defined the requirement for p53 function in numerous tissues, as without Trp53, various tumors rapidly killed p53-deficient animals.16, 17 Mice deficient in one or both Trp53 alleles were susceptible to a spectrum of tumor types including brain and lung, lymphomas and sarcomas, and also exhibited resistance to several inducers of DNA damage.17, 18
More recently, several groups have honed genetic approaches to directly test p53 mutations similar to those found in Li–Fraumeni Syndrome patients (by creating a murine knock-in of the mutation) to better understand the biology of naturally occurring, disease-causing p53 mutations.19, 20 The p53 point mutations selected were Trp53R172H (corresponding to Trp53R175H in humans) and Trp53R270H (corresponding to Trp53R273H in humans), which are structural and DNA contact mutants, respectively. The phenotypes of the Trp53R172H/+ and Trp53R270H/+ knock-in animals demonstrated that there are dramatic differences compared to Trp53+/−. One impressive difference is the tumor profile that developed in these models. A distinct set of tumors, such as B-cell lymphomas occurred in both the Trp53R172H/+ and Trp53R270H/+ animals, and a marked increase of carcinomas, adenomas, hemangiomas, and osteocarcomas compared to Trp53+/−. Also striking, is that Trp53R172H/+ tumors demonstrated a greater propensity to metastasize, which was corroborated in a similar study. The proposed mechanisms responsible for these differences center on mutant forms of p53 acquiring a ‘gain of function’ to cooperate with p53wt and alter, perhaps, protein–protein interactions between p53 and it's binding partners, which could lead to changes in transcriptional regulation and stability.
While the models described above support the idea that loss of p53 function is deleterious to maintaining a tumor-free state; they also suggest that it will be difficult to ascertain which function(s) of p53 is primarily responsible for maintaining tumor suppression. Even so, several elegant studies have proposed that p53-dependent apoptosis is the major function required for tumor suppression in vivo.21, 22 Several years ago it was shown that a decrease in p53-induced apoptosis correlated with the development of aggressive tumor emergence. A more recent study has highlighted the importance of p53's ability to induce apoptosis in preserving a tumor-free state.22 In a model of B-cell lymphoma induced by the enforced expression of the Eμ-myc transgene, spontaneous Trp53 mutations arose; and likewise, a marked acceleration of tumor onset occurred when p53 function was inactivated.23 The pressure to inactivate p53 function in this model was substantially reduced when Eμ-myc-expressing tumors were infected with retroviruses encoding Bcl-2 or dominant-negative caspase-9 (C9DN).22 Both of these proteins significantly abrogate the induction of apoptosis by either directly interfering with MOMP or reducing the activation of caspases, respectively. Since neither of these proteins interfered with the induction of cell-cycle arrest by p53, it was hypothesized that expression of Bcl-2 or C9DN is sufficient to inhibit p53-induced death, and allowed for tumor progression. Effectively, expression of Bcl-2 or C9DN phenocopied the loss of p53 in this model system, suggesting that apoptosis is the critical p53 tumor suppressor function.
Although it is clear that p53 is an important tumor suppressor, some evidence suggests that apoptosis may not be the keystone to inhibiting tumor formation. Data from the SWAP mouse (the human Trp53 gene is knocked-into the murine Trp53 locus) indicated that while the SWAPed p53 followed normal regulation by ultraviolet light and gamma-irradiation, in terms of phosphorylation and stabilization, it failed to induce apoptosis (and cell-cycle arrest) in several tumorigenesis models (Dmitry Bulavin, personal communication). However, the SWAPed p53 could rescue the early onset of death in Trp53−/− animals and substantially restore abnormal centrosomal checkpoint, indicating that non-apoptotic functions may assist in cellular stability. Interestingly, the SWAPed p53 failed to trans-activate several pro-apoptotic genes, but demonstrated normal trans-repression following ionizing irradiation; this hinted that protein–protein interactions or trans-repression may be essential for regulating tumor formation. In models of lymphomagenesis, the SWAPed p53 was often lost indicating that this form of p53 indeed had produced a selective pressure to tumor formation. Yet, it can be difficult to grasp the impact of human p53 in the context of a foreign signaling circuit, as the SWAPed p53 could possess gain- or loss-of-function behaviors in the murine milieu.
One must appreciate the complexity of p53-induced apoptosis, and understand that all p53-regulated cell death pathways are not equal. This notion is highlighted by the trans-activation-deficient double mutant L25Q, W26S Trp53 knock-in (referred to as p53QS).24, 25 Mouse embryonic fibroblasts derived from the p53+/QS animal demonstrated both compromised trans-activation of numerous p53-induced genes and cell-cycle G1 checkpoint activation. Yet, the induction of p53-regulated apoptosis was stress-dependent: DNA-damaged cell death was completely compromised, while hypoxia-initiated apoptosis was almost similar to controls. So what makes the difference? Perhaps the difference between the two stimuli is the required p53 function in each scenario. DNA-damaged induced cell death required trans-activation of pro-apoptotic genes (other than Bax, as this gene maintained regulation by p53QS), while the hypoxia-induced cascade requires alternative p53 functions, such as those described as transcription-independent. Alternatively, the promoters of pro-apoptotic genes required for a hypoxia response require a different set of protein–protein interactions that are unaffected by the p53QS substitution, and the embryonic lethality of the p53QS/QS animals may support this hypothesis.
The significance of p53-mediated apoptosis in maintaining a tumor-free state may be highlighted by the conserved function of p53 (and it's appropriate homologues) in non-vertebrate model systems. Specifically, the p53 protein in other animals (at least as described in Drosophila and C. elegans) is a pro-apoptotic factor required for a proper DNA-damage response that does not appear to regulate cell cycle.26, 27, 28, 29 Perhaps the adoption of cell-cycle regulation and senescence activities speak to the complexity of vertebrate p53 as these additional functions may contribute to regulating the DNA-damage-induced cell death response in vertebrates.
p53 and Bcl-2 Proteins: Understanding Transcriptional Targets and Protein Interactions
When p53 was described as a pro-apoptotic transcription factor, a commitment to discover the gene(s) responsible for death was imperative. Considerable effort focused on understanding which members of the pro-apoptotic Bcl-2 proteins were regulated by p53, and, not surprisingly, works describing many Bcl-2 proteins and their regulation by p53 can be found. Bax was the first pro-apoptotic Bcl-2 protein described to be upregulated by p53 stabilization following DNA damage, and others, like the BH3-only protein Bid, soon followed.7, 10 Yet, many of these pro-apoptotic molecules are either constitutively expressed in cells (e.g., Bax) or require an additional step to be active (i.e., Bid requires caspase cleavage to be pro-apoptotic). Therefore, the direct impact of exogenous overexpression to mimic p53 upregulation confers limited interpretation as this likely upsets the balance of Bcl-2 proteins rather than reflecting the physiology of each protein. A summary of the fundamental interactions between p53 and various Bcl-2 proteins is presented in Figure 3.

The relationship between p53 and Bcl-2 proteins at the mitochondrial membrane. p53 can either induce the expression of pro-apoptotic Bcl-2 proteins (e.g., Bax, Puma, Noxa), or it can directly regulate numerous Bcl-2 proteins in the cytoplasm. Bax that is expressed by p53 can then be activated by numerous signals, such as p53-induced BH3-only proteins (such as tBid), or by p53 itself. Bax or Bak that is activated by p53 or a BH3-only protein oligomerizes in the mitochondrial outer membrane to allow for MOMP. When Bcl-xL or Bcl-2 is bound by p53, BH3-only proteins once associated with either anti-apoptotic molecule may then be released to either directly activate a pro-apoptotic multi-domain or ‘inhibit’ an additional anti-apoptotic Bcl-2 protein. If nuclear p53 induces the expression of additional BH3-only proteins when cytoplasmic p53 is associated with Bcl-xL, the cell may be sensitized to numerous inducers of death. Alternatively, p53 that is sequestered by Bcl-xL may be liberated by a collateral signal, such as Puma, which frees p53 to activate Bax. Red and green arrows indicate transcriptional and cytoplasmic regulation, respectively
As the pro-apoptotic subfamily of Bcl-2 proteins has increased to include more BH3-only proteins, further evidence suggested that these proteins also exhibit marked p53 regulation. Two such p53 transcriptional targets are NOXA and PUMA, which were originally characterized as BH3-only proteins that directly induce cell death.8, 9 Following p53 stabilization, these genes are transcriptionally upregulated and the proteins were hypothesized to directly induce MOMP. Yet, the evidence that this was indeed correct was based only on overexpression of each protein in cell lines. Further studies demonstrated that the Noxa and Puma BH3 domain peptides did not directly activate recombinant Bax or permeabilize mammalian mitochondria and Puma protein could not directly induce MOMP.30, 31
Genetic evidence from several groups, however, did propose that NOXA and PUMA were important mediators of p53-induced apoptosis.32, 33, 34 NOXA -deficient mice developed normally and exhibited proper cellularity of the hemopoietic organs, but the MEFs were slightly resistant to etoposide-induced cell death.32, 33 The most dramatic results were obtained from oncogene-expressing MEFs which displayed marked resistance to p53-dependent, UV-induced apoptosis.33 In vivo, the absence of NOXA resulted in resistance to X-ray-induced apoptosis in the jejunum and small intestinal crypts.32 The selective resistance to p53-induced apoptosis by the loss of NOXA cannot be explained by the simple explanation that BH3-only proteins can substitute for each other in apoptotic pathways, but rather these proteins function in distinct signaling cascades.30, 35 Noxa is likely to participate in p53-mediated signaling, but the exact pathways and the consequences of its activity are still unresolved.
The absence of PUMA, however, resulted in a broader resistance to numerous p53-dependent and -independent death stimuli in vivo, indicating that this protein likely functions in numerous apoptotic networks.33, 34 Like the NOXA−/− animals, PUMA−/− animals were born at the expected Mendelian frequency, appeared normal and exhibited proper cellularity of the hemopoietic organs.33 Yet, numerous PUMA−/− cell types (e.g., thymocytes, pre-B and mature B- and T-cells) were markedly resistant to inducers of genotoxic stress (e.g., γ-irradiation); but not to the same level as Trp53−/− suggesting that the signaling cascade Puma regulates may be alternatively modulated by other proteins or mechanisms.33 PUMA −/− MEFs expressing the E1A oncogene were also resistant to etoposide-induced apoptosis, similar to NOXA−/− MEFs, suggesting that functional redundancy may not be evident under this condition. Puma may also regulate p53-independent pathways because CD4+8+ thymocytes derived from PUMA−/− animals displayed slight resistance to dexamethasone and staurosporine.33 This suggests that Puma may participate in several death networks to sensitize cells to death stimuli that are regulated by diverse pro- and anti-apoptotic Bcl-2 proteins. Indeed, recent in vitro evidence supports this notion and suggests that the BH3-only proteins may function in distinct signaling networks;30, 31, 35 furthermore, in vivo data from the NOXA and PUMA double knockout (NOXA−/−/PUMA−/−) present no synergistic effect in regard to DNA-damage sensitivity (Andreas Strasser, personal communication).
While the genes encoding the Bcl-2 proteins are transcriptionally regulated by p53, the influence of p53 on this protein family's function does not end in the nucleus. It is now well established that p53 also accumulates in the cytoplasm following stabilization.36, 37, 38, 39, 40, 41 The accumulation of p53 into the cytosol seems to be regulated by Mdm2, as polymorphic variants of p53 that interact better with Mdm2 accumulate more in the cytoplasm.38 Forced localization of p53 to mitochondria can directly induce MOMP, and this is blocked by the co-expression of anti-apoptotic BCL-2 proteins.36, 38, 42 This inhibition proposed that p53's ability to induce MOMP was directly regulated by pro-apoptotic Bcl-2 proteins. Likewise, several groups showed that p53 can interact with Bcl-2 and Bcl-xL, which explains how p53 can bind to mitochondria, contrary to early studies that demonstrated p53 associated with mitochondrial chaperones.36, 37 More recently, endogenous p53 has been shown to interact with Bcl-xL, and because Bcl-xL can be localized on mitochondria in several tissues, endogenous p53 can also be found on mitochondria via a Bcl-xL interaction (or, mitochondrial Bcl-2 and Mcl-1 as well).31, 43 The ability of endogenous p53 to rapidly accumulate (i.e., within 30 min of treatment) in the cytoplasm of radiosensitive organs following γ-irradiation has also been demonstrated in vivo in mice.44 Most studies exploring the association of p53 with anti-apoptotic Bcl-2 proteins focused on Bcl-xL, which also led to a NMR solution structure of p53 and Bcl-xL.45 Although, Bcl-2 almost certainly participates in this pathway as Bcl-2 overexpression also blocks p53-induced death, p53 can bind to Bcl-2, and silencing BCL-2 by RNAi has been shown to induce p53-dependent apoptosis in certain systems.46
One major issue focused on determining the significance of the signal that the p53/Bcl-xL association creates within the cell. Is this the signal to initiate apoptosis, or is a collateral signal required? To examine this, a BH3-binding mutant of Bcl-xL (Bcl-xLG138A) was tested for it's ability to bind p53 and either inhibit or promote cell death.31 Interestingly, Bcl-xLG138A efficiently bound p53 and inhibited p53-induced death. Moreover, when BCL-XL −/− MEFs were reconstituted to stably express Bcl-xLG138A p53-induced apoptosis was also inhibited. This result led the investigators to hypothesize that the binding of p53 to Bcl-xL was not the death signal, and perhaps required an additional signal, such as a p53-regulated BH3-only protein (as it could then bind to Bcl-xL), to promote apoptosis. Furthermore, keeping in mind the requirement for Puma in p53-mediated death, it was speculated that perhaps Puma released p53 from Bcl-xL. Indeed, the Puma BH3 domain peptide and protein acted very specifically to disrupt the p53/Bcl-xL association, and cells that expressed Bcl-xLG138A, while were efficient at holding p53 inactive, failed to undergo cell death because Puma could not bind to Bcl-xLG138A.31
However, what role does p53 play once liberated from Bcl-xL? Data suggest that p53 can act similarly to an activator BH3 protein (e.g., tBid or Bim) to directly induce the oligomerization of pro-apoptotic multi-domain Bcl-2 proteins; this function of p53 has been described for both Bak and Bax.37, 43 The addition of p53 to isolated mitochondria caused MOMP and the release of cytochrome c. Furthermore, in a liposome-based system designed to faithfully mimic permeabilization of the mitochondrial outer membrane, p53 functioned similar to tBid to induce the oligomization and pore-forming activity of purified, recombinant Bax.37, 47 It is worthy to note that the binding of p53 to Bcl-xL (which would repress Bcl-xL's anti-apoptotic function) may also serve to sensitize cells to further stresses that would result in BH3-only protein activation (as they would not be sequestered by p53-bound Bcl-xL).
The domains of p53 responsible for the interaction with Bcl-xL and subsequent MOMP are still not fully understood. Evidence suggests that both the DNA-binding domain (amino acids ∼100–300) and the proline-rich domain (amino acids ∼60–90) are essential for these interactions and cell death.36, 37, 45, 48 Interestingly, short peptides derived from the core domain of p53 exhibited the ability to activate multi-domains, but further work is required (Maureen Murphy, personal communication). The ability of p53 to translocate to the cytoplasm and mitochondria is regulated by MDM2 binding, and it has been shown that polymorphic variants of p53 possess distinct pro-apoptotic potentials based on their ability to be translocated out of the nucleus,38
The interactions described above are relatively recent observations, and the complexity of how other proteins that bind p53 and Bcl-2 proteins, including the myriad post-translational modifications each protein may demonstrate (e.g., ASPPs), impact on p53's pro-apoptotic function have not yet been elucidated.
Making the Decision to Die: Regulation of p53-dependent Apoptosis
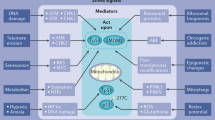
If we consider that MOMP is central to the initiation of apoptosis, then the actual signaling mechanism(s) of p53-induced apoptosis is what lies between p53 stabilization and permeabilization of mitochondria (as the permeabilization of mitochondrial membranes, cytochrome c release and caspase activation are not unique to the p53-dependent pathway). In general, many of the proteins shown to promote or inhibit p53-dependent pro-apoptotic signaling have been only described to regulate p53's nuclear activity, that is, p53-dependent gene expression. Yet, as the extranuclear functions of p53 are being elucidated, several of these proteins may directly regulate p53 in the cytoplasm. Independent of cellular localization, apoptotic signaling is a very tangled network; the modulators of p53-dependent apoptosis are numerous and are themselves subject to regulation. As this area of research is so immense, this section will only highlight the topics discussed at the p53 workshop (Figure 4).

Cellular and pharmacological regulators of the p53-dependent apoptotic pathway. Understanding what is between p53 stabilization and MOMP/apoptosis is not well known. Most distal to the death signal is the relationship between p53 and MDM2. Until the interactions between these proteins are disrupted, no p53 signal can be induced. Data suggest that various proteins (YB1, CHC, ASPP1/2) cooperate with p53 to appropriately target pro-apoptotic genes and dictate the decision to die. Likewise, proteins like Pidd contribute in the p53 signaling cascade by bridging additional pathways to ensure death. Already mentioned in Figures 2 and 3 are the important interactions between p53 and Bcl-2 proteins. The blue dotted arrow indicates potential interactions with the nuclear and cytoplasmic functions of p53. The indicated small molecules demonstrate the different levels of p53 signaling that are currently regulated by pharmacological means. The interactions between p53 and Mdm2 (RITA and the Nutlins), altering mutant forms of p53 to behave like wild-type p53 (PRIMA-1) and inhibitors to p53-induced gene regulation (pifithrin-α) all hold promise, at least in theory, as ‘druggable targets’ that will hopefully benefit those receiving anti-tumor therapies
Once p53 is stabilized and accumulates in the nucleus, it must target a specific repertoire of pro-apoptotic genes to induce cell death. To achieve this, p53 either becomes post-translationally modified to recognize specific promoters or it cooperates with transcriptional regulators that deliver p53 to the appropriate site (most likely, both happen). Commonly, p53 induces the upregulation of pro-apoptotic genes, such as BAX and PUMA,7, 9 yet under conditions where cellular machinery may be compromised, for example, hypoxia, p53 can repress the expression of anti-apoptotic genes, such as survivin which may promote caspase activation.49, 50 The function of p53 to repress anti-apoptotic genes targets numerous gene families, such as BCL-2; loss of BCL-2, BCL-xL or MCL-1 expression would sensitize a cell apoptosis by de-repressing activated BH3-only proteins (e.g., Bim). Another scenario demonstrating the importance of repression in p53-induced death is highlighted by recent evidence that wild-type Trp53-expressing thyroid papillary carcinomas51 fail to induce p53-dependent apoptosis owing to a loss of Galectin-3 downregulation. This is perhaps owing to the lack of coordinated p53 and homeodomain-interacting protein kinase 2 (HIPK2) trans-repression of Galectin-3, an anti-apoptotic factor, potentially by a loss of heterozygosity for the HIPK2 gene (Silvia Soddu, personal communication).
As mentioned earlier, p53 also coordinates with other factors to selectively target pro-apoptotic gene expression. Y-box binding protein-1 (YB1) was recently shown to block p53-dependent apoptosis, and not cell-cycle arrest or MDM2 activity, by specifically abrogating BAX expression; these data support the notion that YB1 expression is associated with poor prognosis.52 On the contrary, clathrin heavy chain (CHC), has been described to interact with p53 and enhance p53 reporter activity and apoptosis (Yoichi Taya, personal communication). Yet, CHC is a well-described cytosolic protein that regulates vesicle transport during endocytosis. Even though CHC has been shown to interact with p53 on p53 response elements by chromatin immunoprecipitation analysis in vitro, CHC may potentially regulate p53's cytoplasmic function as well.
As the transcriptional regulation of Bcl-2 proteins alone may not be sufficient to induce MOMP under all circumstances, p53 also induces the genes for pro-apoptotic proteins that may link intrinsic and extrinsic apoptotic programs. One example is p53-indicible death domain (PIDD), the protein of which contains a series of seven leucine rich repeats and a death domain.53, 54 These motifs are generally responsible for protein–protein interactions and transmit a cell death signal. Exogenous PIDD has been shown to induce apoptosis, whereas the loss of PIDD expression abrogated p53-dependent death. The mechanism of Pidd-induced death is not well established, but may involve caspases-2, -8, APAF-1 (apoptotic protease activation factor-1), FADD (Fas-associated death domain) and RAIDD (RIP-associated ICH-1/CED-3 homologous protein with a death domain).53 It is described that PIDD, RAIDD and caspase-2 association resulted in caspase-2 activation,55 but the impact of caspase-2 in p53-induced apoptosis is not concrete, as CASPASE-2 deficiency does not abrogate p53 responses in vivo.
A promising area of study involves the family of ASPP (ankyrin repeat, SH3 domain and proline-rich domain containing proteins/apoptosis stimulating proteins of p53) proteins that are comprised of both positive (ASPP1 and ASPP2) and negative effectors (iASPP, an ASPP1/2 inhibitor)56 (see review by Braithwaite et al. for further discussions). ASPP1 or ASPP2 coordinated with p53 to increase p53-dependent apoptosis, but they have no ability to enhance p53-dependent cell-cycle arrest.57 In contrast, iASPP specifically inhibited p53-dependent apoptosis and cooperated with Ras, E1A or E7 to transform cells in vitro.58 The mechanism of ASPP1/2 action was suggested to enhance the ability of p53 to trans-activate pro-apoptotic promoters, and iASPP could block this effect (interestingly, anti-sense iASPP promoted p53-dependent apoptosis, suggesting this mechanism is constitutively engaged). Yet, it is fairly common that p53 target genes (e.g., cell cycle and apoptosis) are simultaneously induced, and therefore the mechanism by which ASPP1/2 specifically enhanced pro-apoptotic promoter activity is not known. ASPP2, however, can also associate with numerous other proteins, such as Bcl-2.59 The result of this interaction is hypothesized to inhibit Bcl-2 function and promote p53-dependent apoptosis (although loss of Bcl-2 alone does not do this). One additional possibility is perhaps that the Bcl-2/ASPP2 interaction regulates the cytoplasmic localization and activity of p53.
Waking up the Guardian: p53-induced Apoptosis as a Drug Target
Evidence that p53 is a promising pharmacological target (see review by Klaus Wiman in this issue for a detailed discussion of this topic) surfaced when peptides derived from the C-terminal region of p53 restored mutant p53 function and allowed for p53-dependent apoptosis.60 Small molecules that corrected the conformation of mutant p53 soon followed, but the efficiency and required doses were not suitable for clinical use.61 This was potentially due to the drug having to remodel the p53 protein after translation. However, these drugs rapidly evolved and another drug that could reactivate mutant p53 (both DNA contact and structural mutants – by restoring the wild-type conformation to the DNA-binding domain) to regulate p53-dependent gene expression and apoptosis was described. That drug, PRIMA-1 (p53 reactivation and induction of massive apoptosis) was hypothesized to bind nascent mutant p53 protein, rather than having to restore function to protein already folded, thus lowering the required dose (an additional drug, MIRA-1 has also been described to provoke a wild-type conformation in mutant p53 species) (Figure 4).62 The mechanism of action for each of these drugs has not been elucidated beyond the requirement for mutant p53 and its ability to bind to p53 response elements. However, we can extend the data mentioned in earlier sections (e.g., the treated mutant p53 protein can now trans-activate the appropriate pro-apoptotic genes, bind Bcl-2 proteins, induce MOMP and caspase activation) to grasp how these molecules trigger cell death. Yet, since cells that harbor mutant p53 often exhibit marked hyper-expression and mis-localization of p53, the actual mechanism of action cannot be assumed. These caveats may, however, contribute to the observed death in tumor models, as the drugs appear to induce apoptosis in the absence of genotoxic stress (which would normally be required to stabilize the p53 protein).
The requirement for genotoxic stress or other p53-stabilizing stimuli can also be overcome by inhibiting the interaction between p53 and Mdm2, thus allowing p53 to accumulate (as the Trp53 promoter is almost constitutively active) to promote apoptosis. One molecule, RITA (reactivation of p53 and induction of tumor cell apoptosis), was described to induce the expression of p53 target genes and marked apoptosis in wild-type p53-expressing cell lines and could coordinate an anti-tumor response upon oncogene expression.63 RITA functioned by abrogating the p53/Mdm2 ·MDM2 interaction via binding the N-terminus of p53. A family of small molecules aimed at targeting the p53 binding domain of Mdm2, the Nutlins, were also able to induce robust p53-dependent apoptosis in tumor xenograft models.64 Again, this class of molecules that target the p53/Mdm2·interaction display pro-apoptotic function due to the activities of p53 described earlier (if these drugs are truly specific to the p53 pathway). This is exemplified by chromatin condensation detected by TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling) staining and nuclear morphology.
These small molecules are also attractive because they can engage p53 activity in the presence of hyper-expressed Mdm2 or other situations where the p53/Mdm2· interaction is pathologically promoted.65, 66 The Nutlins, in particular, provide both in vitro and in vivo ‘proof-of-principle’ evidence that the p53/Mdm2· interface is a key drug target. As a note, an inhibitor of p53-induced apoptosis has also been described, pifithrin-α.67 This drug may protect non-tumorigenic tissues from the undesirable side effects of cancer treatments.
Conclusion
There are a few messages that emerge from our understanding of p53-induced apoptosis. First, the ability of p53 to trigger cell death is a key tumor suppressor activity. Second, the cellular signaling cascades leading to p53-dependent apoptosis are numerous, perhaps specific to the inducer, and probably not linear (i.e., one p53 trans-activated gene may not be responsible for the entire p53 death circuit). Finally, the p53-regulated apoptotic pathway can be pharmacologically modulated to hopefully treat cancer patients. Taking these three ideas into consideration will help us to reach a better understanding of the relevance of p53 and cell death in both principle and practice.
Abbreviations
- APAF1:
-
apoptotic protease activating factor 1
- ASPP:
-
ankyrin repeat, SH3 domain and proline-rich domain containing proteins/apoptosis stimulating proteins of p53
- BAK:
-
Bcl-2 antagonist/killer
- BAX:
-
Bcl-2-associated X protein
- BH3:
-
Bcl-2-homology domain-3
- BID:
-
BH3-interacting-domain
- ced-3 :
-
Caenorhabditis elegans cell-death abnormality-3
- C9DN:
-
dominant-negative caspase-9
- CHC:
-
clathrin heavy chain
- FADD:
-
FAS-associated death domain
- HIPK2:
-
homeodomain-interacting protein kinase 2
- iASPP:
-
inhibitor of ASPP1/2
- ICE:
-
interleukin-1 converting enzyme
- MCL-1:
-
myeloid cell leukemia-1
- MOMP:
-
mitochondrial outer-membrane permeabilization
- p53 QS :
-
L25Q, W26S Trp53 knock-in
- PIDD :
-
p53-indicible death domain
- PRIMA-1:
-
p53 reactivation and induction of massive apoptosis
- PUMA:
-
p53-upregulated modulator of apoptosis
- RITA:
-
reactivation of p53 and induction of tumor cell apoptosis
- tBID:
-
truncated BID
- TNF:
-
tumor necrosis factor
- TNFR1:
-
TNF receptor 1
- TRADD:
-
TNFR1-associated death domain
- TUNEL:
-
terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling
- YB1:
-
Y-box binding protein-1
References
Braithwaite AW, Royds JA and Jackson P (2005) The p53 story: layers of complexity. Carcinogenesis 26: 1161–1169.
Danial NN and Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219.
Spierings D, McStay G, Saleh M, Bender C, Chipuk J, Maurer U and Green DR (2005) Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science 310: 66–67.
Muppidi JR, Tschopp J and Siegel RM (2004) Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 21: 461–465.
Vogelstein B, Lane D and Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310.
Xu Y (2003) Regulation of p53 responses by post-translational modifications. Cell Death Differ. 10: 400–403.
Miyashita T and Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T and Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058.
Nakano K and Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7: 683–694.
Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ and El-Deiry WS (2002) BID regulation by p53 contributes to chemosensitivity. Nat. Cell Biol. 4: 842–849.
Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M and Krammer PH (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 188: 2033–2045.
Moroni MC, Hickman ES, Lazzerini Denchi E, Caprara G, Colli E, Cecconi F, Muller H and Helin K (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 3: 552–558.
Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y and Taya Y (2000) p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102: 849–862.
Wu GS, Burns TF, McDonald III ER, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, We G and El-Deiry WS (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 17: 141–143.
Schuler M and Green DR (2005) Transcription, apoptosis and p53: catch-22. Trends Genet. 21: 182–187.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery Jr CA, Butel JS and Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221.
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT and Weinburg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4: 1–7.
Harvey M, McArthur MJ, Montgomery Jr CA, Butel JS, Bradley A and Donehower LA (1993) Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat. Genet. 5: 225–229.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK and Lozano G (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li–Fraumeni syndrome. Cell 119: 861–872.
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D and Jacks T (2004) Mutant p53 gain of function in two mouse models of Li–Fraumeni syndrome. Cell 119: 847–860.
Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T and Van Dyke T (1994) p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell 78: 703–711.
Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM and Lowe SW (2002) Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 1: 289–298.
Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD and Brinster RL (1985) The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318: 533–538.
Johnson TM and Attardi LD (2005) p53QS: an old mutant teaches us new tricks. Cell Cycle 4: 731–734.
Johnson TM, Hammond EM, Giaccia A and Attardi LD (2005) The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat. Genet. 37: 145–152.
Derry WB, Putzke AP and Rothman JH (2001) Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294: 591–595.
Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM and Abrams JM (2000) Drosophila p53 binds a damage response element at the reaper locus. Cell 101: 103–113.
Jin S, Martinek S, Joo WS, Wortman JR, Mirkovic N, Sali A, Yandell MD, Pavletich NP, Young MW and Levine AJ (2000) Identification and characterization of a p53 homologue in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 97: 7301–7306.
Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaken K, Demsky M, Fisher WW, Buchman A, Duy KG, Friedman L, Prives C and Kopczynski C (2000) Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101: 91–101.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR and Newmeyer DD (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17: 525–535.
Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD and Green DR (2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309: 1732–1735.
Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T and Tanaka N (2003) Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17: 2233–2238.
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM and Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038.
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL and Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM and Huang DC (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17: 393–403.
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P and Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 11: 577–590.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M and Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014.
Dumont P, Leu JI, Della Pietra III AC, George DL and Murphy M (2003) The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 33: 357–365.
Giannakakou P, Sackett DL, Ward Y, Webster KR, Blagosklonny MV and Fojo T (2000) p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat. Cell Biol. 2: 709–717.
Lomonosova E, Subramanian T and Chinnadurai G (2005) Mitochondrial localization of p53 during adenovirus infection and regulation of its activity by E1B-19K. Oncogene 24: 6796–6808.
Tan J, Zhuang L, Leong HS, Iyer NG, Liu ET and Yu Q (2005) Pharmacologic modulation of glycogen synthase kinase-3beta promotes p53-dependent apoptosis through a direct Bax-mediated mitochondrial pathway in colorectal cancer cells. Cancer Res. 65: 9012–9020.
Marchenko ND, Zaika A and Moll UM (2000) Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 275: 16202–16212.
Leu JI, Dumont P, Hafey M, Murphy ME and George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 6: 443–450.
Erster S, Mihara M, Kim RH, Petrenko O and Moll UM (2004) In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol. Cell. Biol. 24: 6728–6741.
Petros AM, Gunasekera A, Xu N, Olejniczak ET and Fesik SW (2004) Defining the p53 DNA-binding domain/Bcl-x(L)-binding interface using NMR. FEBS Lett. 559: 171–174.
Jiang M and Milner J (2003) Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev. 17: 832–837.
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR and Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342.
Chipuk JE, Maurer U, Green DR and Schuler M (2003) Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 4: 371–381.
Hoffman WH, Biade S, Zilfou JT, Chen J and Murphy M (2002) Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 277: 3247–3257.
Hammond EM and Giaccia AJ (2005) The role of p53 in hypoxia-induced apoptosis. Biochem. Biophys. Res. Commun. 331: 718–725.
Moore D, Ohene-Fianko D, Garcia B and Chakrabarti S (1998) Apoptosis in thyroid neoplasms: relationship with p53 and bcl-2 expression. Histopathology 32: 35–42.
Homer C, Knight DA, Hananeia L, Sheard P, Risk J, Lasham A, Royds JA and Braithwaite AW (2005) Y-box factor YB1 controls p53 apoptotic function. Oncogene 24: 8314–8325.
Berube C, Boucher LM, Ma W, Wakeham A, Salmena L, Hakem R, Yeh WC, Mak TW and Benchimol S (2005) Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc. Natl. Acad. Sci. USA 102: 14314–14320.
Lin Y, Ma W and Benchimol S (2000) Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat. Genet. 26: 122–127.
Tinel A and Tschopp J (2004) The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304: 843–846.
Slee EA and Lu X (2003) The ASPP family: deciding between life and death after DNA damage. Toxicol. Lett. 139: 81–87.
Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T and Lu X (2001) ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 8: 781–794.
Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK, O'Connor DJ, Zhong S, Campargue I, Tomlinson ML, Kuwabara PE and Lu X (2003) iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat. Genet. 33: 162–167.
Naumovski L and Cleary ML (1996) The p53-binding protein 53BP2 also interacts with Bc12 and impedes cell cycle progression at G2/M. Mol. Cell. Biol. 16: 3884–3892.
Selivanova G, Ryabchenko L, Jansson E, Iotsova V and Wiman KG (1999) Reactivation of mutant p53 through interaction of a C-terminal peptide with the core domain. Mol. Cell. Biol. 19: 3395–3402.
Foster BA, Coffey HA, Morin MJ and Rastinejad F (1999) Pharmacological rescue of mutant p53 conformation and function. Science 286: 2507–2510.
Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG and Selivanova G (2002) Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 8: 282–288.
Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A and Selivanova G (2004) Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 10: 1321–1328.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N and Liu EA (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848.
Vassilev LT (2004) Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle 3: 419–421.
Vassilev LT (2005) p53 Activation by small molecules: application in oncology. J. Med. Chem. 48: 4491–4499.
Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV and Gudkov AV (1999) A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285: 1733–1737.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by A Braithwaite
Rights and permissions
About this article
Cite this article
Chipuk, J., Green, D. Dissecting p53-dependent apoptosis. Cell Death Differ 13, 994–1002 (2006). https://doi.org/10.1038/sj.cdd.4401908
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401908
Keywords
This article is cited by
-
Multipronged regulation of autophagy and apoptosis: emerging role of TRIM proteins
Cellular & Molecular Biology Letters (2024)
-
The expression level of ARF and p53 in AML patients, and their relation to patients' outcome
Egyptian Journal of Medical Human Genetics (2023)
-
Time-dependent unloading effects on muscle and bone and involvement of FNDC5/irisin axis
npj Microgravity (2023)
-
Exploring the multiple roles of guardian of the genome: P53
Egyptian Journal of Medical Human Genetics (2020)
-
The Localization of p53 in the Crayfish Mechanoreceptor Neurons and Its Role in Axotomy-Induced Death of Satellite Glial Cells Remote from the Axon Transection Site
Journal of Molecular Neuroscience (2020)
