
The Coming of Age of the Angiotensin Hypothesis in Alzheimer’s Disease: Progress Toward Disease Prevention and Treatment?
Abstract
There is wide recognition of a complex association between midlife hypertension and cardiovascular disease and later development of Alzheimer’s disease (AD) and cognitive impairment. While significant progress has been made in reducing rates of mortality and morbidity due to cardiovascular disease over the last thirty years, progress towards effective treatments for AD has been slower. Despite the known association between hypertension and dementia, research into each disease has largely been undertaken in parallel and independently. Yet over the last decade and a half, the emergence of converging findings from pre-clinical and clinical research has shown how the renin angiotensin system (RAS), which is very important in blood pressure regulation and cardiovascular disease, warrants careful consideration in the pathogenesis of AD. Numerous components of the RAS have now been found to be altered in AD such that the multifunctional and potent vasoconstrictor angiotensin II, and similarly acting angiotensin III, are greatly altered at the expense of other RAS signaling peptides considered to contribute to neuronal and cognitive function. Collectively these changes may contribute to many of the neuropathological hallmarks of AD, as well as observed progressive deficiencies in cognitive function, while also linking elements of a number of the proposed hypotheses for the cause of AD. This review discusses the emergence of the RAS and its likely importance in AD, not only because of the multiple facets of its involvement, but also perhaps fortuitously because of the ready availability of numerous RAS-acting drugs, that could be repurposed as interventions in AD.
INTRODUCTION
As Journal of Alzheimer's Disease celebrates its 20th anniversary, this timeframe has also seen the emergence of research that points strongly to the involvement of the renin angiotensin system (RAS) as a likely, fortunately already modifiable, factor in the development and pathogenesis of Alzheimer’s disease (AD; MIM 104300 (https://www.omim.org/entry/104300)). While AD represents the most common form of dementia, with characteristic neuropathological hallmarks, it exists alongside a number of other causes of dementia, that have overlapping or related neuropathological processes and hallmarks. Yet, all of the causes of the various dementias still share the same damning lack of therapeutic options, that are now vital to address the ongoing and escalating health care crisis that dementia presents in an increasingly aging population [1].
A large proportion of people diagnosed with AD have concurrent cerebrovascular disease (CVD) of variable severity, alongside the widely known characteristic AD-related amyloid-β (Aβ) pathologies like senile plaques and cerebral amyloid angiopathy (CAA), as well as tau-protein related neurofibrillary tangle pathology [2–4]. While AD shares many of the same risk factors for CVD and vascular cognitive impairment, the presence of vascular risk factors or CVD exacerbates the progression, or at least lowers the clinical threshold for the manifestation, of AD [5, 6]. There seems to be a highly intimate and complex temporal relationship between the development of cardiovascular risk factors, CVD, and subsequent development and/or contribution toward the pathogenesis of AD. These may also contribute to age-associated cognitive decline. Embedded within this relationship appear to be mediators of RAS function that are characteristic in blood pressure regulation and cardiovascular diseases like hypertension, but which more recently have been consistently noted to be involved in numerous pathological processes that are present in AD.
This review provides an overview of the emergence of the RAS as a biochemical pathway that can have a chronic and integral role in the development and pathogenesis of AD. From initial hints of involvement in the pre-genome wide association studies (GWAS) era of genetic association studies in AD; through numerous consistently supportive and converging findings to numerous pre-clinical studies, the RAS has rose to some prominence. The concurrent emergence of supportive research findings at a population level have also helped to further elevate the RAS, as a mechanism that may explain the widely accepted, but not well understood, association between mid-life hypertension and the development of cognitive impairment and/or dementia later in life. The convergence of genetic, molecular, and epidemiological evidence, and the fortunate availability of numerous drugs that work effectively to inhibit RAS activity, has now brought forth the now very credible evidence that implicates RAS involvement in AD. Fortunately, this line of research can be effectively and rapidly tested, using clinical trials of already available RAS acting drugs, in early and mid-phase clinical trials for AD.
HYPOTHESES OF ALZHEIMER’S DISEASE: THE PARABLE OF THE BLIND MONKS AND THE ELEPHANT
The neuropathological characterization of AD relates to assessment of the presence of intracellular neurofibrillary tangles and extracellular deposition of various isoforms of Aβ in the forms of senile plaques. Another characteristic that is common in AD, but not considered as part of the diagnosis, is the deposition of Aβ in blood vessels in the brain known as CAA [4]. The presence of such features in the postmortem brain tissue, considered alongside a medical history that refers to progressive memory loss and cognitive impairment, all help to provide what currently remains as the only method to obtain a confirmatory diagnosis of AD.
For decades, theories on the development of AD have been based, in no small part, on the amyloid cascade hypothesis and the cholinergic hypothesis. These have both been extensively written about and updated in the intervening years. The amyloid cascade hypothesis [7] describes the significant pathogenic contribution of Aβ peptide, derived from cleavage of the amyloid-β precursor protein (AβPP), and its accumulation in the brain as a result of imbalance between its production and clearance [8–11]. The amyloid cascade hypothesis has been the focus of numerous recent unsuccessful but also ongoing, hopefully more successful, clinical trials of new AD therapies with the ultimate aim of reducing the levels of Aβ in the brain by various approaches (reviewed in [12, 13]). The cholinergic hypothesis [14, 15] describes the loss of the neurotransmitter acetylcholine (ACh) in the central nervous system, a major factor in the progressive cognitive decline associated with AD. The reductions in ACh are linked to reductions in levels of the ACh synthesizing enzyme, choline acetyltransferase (ChAT) and the progressive neuronal loss that is seen in AD [16, 17], which gives rise to reductions in levels of nicotinic and muscarinic ACh receptors (nicotinic (nAChRs) and muscarinic (mAChRs), respectively) [15]. Importantly, most of the licensed drugs currently used to treat some of the symptoms of progressive AD are those that inhibit the breakdown of ACh by acetylcholinesterase, thereby increasing its lifespan. However, these ‘acetylcholinesterase inhibitors’, are not curative since their function is to address the imbalances in ACh and not to modify or halt the progressive neuronal loss that the NMDA receptor antagonist ‘memantine’, as an inhibitor of glutamate signaling, that is released as a consequence of neuronal damage, was originally developed to help alleviate [15]. Ultimately, all of the currently licensed drugs for AD have a limited duration of effect because they are unable to stop the progressive nature of the neuropathology that current anti-Aβ intervention strategies seek to address [12, 13, 18].
Neither hypothesis is complete and self-contained and both have some shortcomings. There is evidence, for example, of positive and negative interactions between elements of the amyloid cascade and cholinergic hypotheses. Some evidence supports a potentially beneficial role of Aβ in regulating the uptake of choline, a vital component in ACh synthesis and degradation, and similarly mediated changes to AChE vesicular ACh transporter (VAChT) proteins to concentrate ACh into the synaptic vesicles from which they are released upon neurotransmission. There are also contrasting reports of the role of Aβ in: inhibiting rapid transport of VAChT; reduced levels and function (including signaling) of receptors of the cholinergic system; and reduced synthesis and release of ACh (for review, see [15]). Other complex and paradoxical interactions include the evidence in animal models of mAChRs influencing the processing of AβPP as well as Aβ-related pathology, whereas activation of nAChRs and nicotine increased cleavage of AβPP by α-secretase to reduce levels of Aβ ([15] for review).
Yet, while these hypotheses are perhaps the most widely known, other hypotheses have also been proposed. A few of these will be summarized given their increasing recognition and support by these examples are not exhaustive. The vascular hypothesis of AD wherein the modification of risk factors of AD and vascular cognitive impairment could prevent, reduce, or delay the onset of any consequential cognitive impairment or dementia [19]. The inflammatory hypothesis of AD seeks to explain how inflammation in response to both Aβ accumulation and tau-related pathology is most likely a major contributor to the progressive neuropathology of AD [20]. The mitochondrial cascade hypothesis proposes that gene inheritance defines an individual’s normal mitochondrial function, which in turn influences rates of change in mitochondrial function over time through interactions with other inherited and environmental factors. These then act together to influence AD chronology including the initiation of any Aβ cascade [21]. Finally, the oxidative stress hypothesis describes numerous links between alterations, some due to genetic variation, in the antioxidant system and increased levels of oxidative damage and mitochondrial disturbances that contribute to the progression of dementia and might be a target for early intervention [22].
There are elements of some of these alternative hypotheses that overlap and that are also consistent with elements of the amyloid cascade and cholinergic hypotheses. As mentioned, the latter has already given rise to some of the current therapies; however, the former, while dominating drug development research in recent decades, has unfortunately yet to deliver a single effective treatment. The failure thus far, of Aβ-targeting interventions, has been suggested by some to be due to over-reliance on considering the Aβ hypothesis as a primary causative process in AD, as a result of misinterpretation of previous findings that were originally presented as evidence in support of the Aβ hypothesis, but that could also be interpreted in a manner that is independent of a role of Aβ in AD [23]. This would also be the interpretation of aspects of some the alternative hypotheses mentioned; however, it must also be noted that the failure to date of Aβ-targeting interventions may not be what is being targeted but when and for how long it is targeted since all end stage clinical trials involve patients with advanced disease with arguably too short a follow-up period. Thus, timing is likely one of the most important factors in the eventual discovery of a new intervention (see below).
On reflection, the various hypotheses proposed for AD echoes with the ancient parable of the blind men and the elephant. This describes a group of blind men attempting to learn about an elephant for the first time by touch and each member of the group proposing an explanation to the others for what it is, based on the individual part of the elephant’s body that they feel. This inevitably gives rise to each explanation being different from the members of the group depending on which body part was felt (e.g., legs, trunk, tail, wall, ears, tusks). Over the last 3 decades, the great complexity of AD has continued to emerge and while fundamental questions remain as to its cause, some comfort should also be taken that there are now a number of hypotheses, a number of which have some degree of overlap with converging elements, and thus collectively will help us gain the complete understanding needed to meet one of the greatest health care challenges of our time.
What continues to be a major stumbling block is the determination of the correct chronology of factors and events that give rise to AD and how these interact at a systems level to explain the progression of the disease and all the neuropathological and clinical nuances that are characteristically seen. The progress thus far provides significant hope for the potential gains to be had from wider adoption of integrative systems biology approaches, that have made substantial contributions to the progress of cancer research, to the study of AD [24]. A wider perspective of the various contributory processes in the pathogenesis of AD is more likely to allow new lines of drug discovery [24], or prompt the reconsideration of the drugs already known and used for other conditions that could be repurposed to have greater benefit in timely studies for the prevention of treatment of AD [25].
DOES TIME HOLD THE KEY FOR THE DEVELOPMENT AND TREATMENT OF ALZHEIMER’S DISEASE?
Apart from the obvious and urgent need to develop treatments for AD, to try and tackle the escalating health care costs associated with the high prevalence in what is an increasingly aged population [1], it has become apparent that ‘timing’ is likely one of the most important factors in achieving success at preventing or effectively treating AD.
It is now widely recognized that the insidious development of AD also involves a lengthy ‘incubation’ period. Indeed by the time typical clinical symptoms of memory loss and cognitive impairment are apparent, there is already advanced disease that could be some decades in development [26]. For a disease that is mainly described as a disease of late onset and predominantly affecting the elderly, for those people who go on to develop the disease, its earliest manifestations that are often described as changes to Aβ biology, which in turn trigger various inflammatory and oxidative mechanisms, could have occurred decades before. Thus for the majority of people that go on to develop AD, what is currently considered ‘middle age’ is likely a crucial time where the brain is at most risk toward the development of the disease [26]. Significant research has been undertaken to identify ways in which people whom might go on to develop AD can be identified as early as possible. This includes efforts to identify biomarkers such as in cerebrospinal fluid (CSF), including the measurement of isoforms Aβ and tau, or magnetic resonance imaging (MRI)-based measures of brain structure and volume, that may be of prognostic value for those still pre-symptomatic but perhaps likely to develop AD [26]. These efforts have been in parallel to thirty years of research to dissect the genetic etiology of AD, where a plethora of risk genes have been suggested, some of which can be used to generate polygenic risk scores, with some reported accuracy to predict whom among people carrying various risk genes, will go on to develop AD [27–29]. However, the genetic contribution to AD still needs to be considered alongside the important influence of lifestyle, diet, and other risk factors as well as the cellular environment in which they function.
It is now clear that epigenetic changes (i.e., modifications to DNA affecting their levels of activity in cells) play a likely role in AD [30], as does the regulation of gene expression by microRNAs [31], the latter field being one still very much in its development. Yet, ours and others’ early pursuits of a better understanding of the genetic etiology of AD yielded the first hints of what might be a role of RAS in AD. This prompted wider investigations that, as a result, has now provided insights into mechanisms that may help to explain the widely known, but poorly understood association between cardiovascular disease and hypertension in particular in mid-life, and the increased risk of developing dementia in later life [32].
HUMBLE AND SOMEWHAT PARADOXICAL BEGINNINGS
Our initial curiosity as to the potential involvement of the RAS in AD arose from our own modest candidate gene association studies in the mid 1990s, in the pre-GWAS era. We sought to test whether variation in the angiotensin I-converting enzyme (ACE) gene (ACE), already implicated in cardiovascular disease [33], might also be associated with susceptibility for AD [34]. We observed a statistically significant and consistent increase in ACE (I) allele bearing genotypes and increased risk of AD, in three independent case-control cohorts, that was independent of any APOE associated risk [34]. This study, which was small by modern standards, but modestly sized in its day, coincided with two other smaller studies that found no evidence of association [35, 36]. In that pre-GWAS era, where underpowered studies were quite common, inconsistent findings were also very common [37]. Yet, unlike many of suggested AD risk genes of that time, the implicated variant (a common Alu (indel) insertion(I)/deletion (D) polymorphism (rs1799752) within intron 16) in ACE, had some functional effect and was already known to influence plasma levels of ACE, the rate-limiting enzyme in the synthesis of the potent vasoconstrictor angiotensin II (ANGII) from angiotensin I (ANGI) [38].
ACE has a complex genetic architecture, being the result of a gene duplication in antiquity but also where tracts of the gene are in very tight association whereby particular polymorphisms that occur have been reported to account for 20% of the total variation in serum ACE concentration and 16–24% of the variation in ACE activity [39–44]. What had been found was that there was a linear association between the lowest plasma ACE levels in ACE I allele homozygotes, through heterozygotes and to D allele homozygotes that were associated with the highest levels of ACE [38]. Yet, there is also evidence that the ACE indel may influence the relative enzymatic contributions of the two (N- and C-) catalytic domains on ACE that give rise to ANGII [45], while others have reported complex negative interactions between the domains that may influence the effectiveness of ACE-inhibitors, that as their name suggests inhibit the activity of ACE and are used to treat hypertension in humans [44].
The existence of functional variants in the gene encoding an enzyme with a fundamental role in blood pressure regulation made ACE a strong candidate gene, and particularly so with additional earlier evidence of altered (increased) activity of ACE in AD in some small postmortem studies [46, 47]. This line of enquiry also fitted well with the earliest inceptions of the vascular hypothesis of AD had been proposed for some years [19]. Over the subsequent decade, numerous replication studies and a number of meta-analyses [48–51], including Alzgene (Gene id = 125 at http://www.alzgene.org/), supported the possible modest involvement of the original variant, and other ACE variants as risk factors for AD [49, 52]. Some studies also reported associations between ACE with earlier ages of onset of AD [53]; smaller hippocampal and amygdalar volumes [54]; and lower (more adverse) levels of CSF Aβ [49]. As the GWAS era evolved, there was also supportive evidence of associations with ACE from family-based and case-control studies [55–59], in association with CSF Aβ levels [60] and of ACE protein level (but not ACE activity) in postmortem CSF from AD patients [61].
ACE currently remains a gene of interest in AD but has not surpassed the stringent significance thresholds currently used to define risk status in more recent GWAS studies [62]. Yet the story of ACE variation in AD has created some confusion. Its original candidacy in AD was based on its potential role as a determinant of vascular effects in AD. However, the risk variants of ACE found to be associated with AD were those normally associated with lower, rather than the higher levels of plasma ACE that was presumed to mediate vascular effects [33]. It was to be a few years before this apparently paradoxical finding might be explained by some unexpected but particularly important data that was to emerge from a series of preclinical investigations.
THE COMPLICATED STORY OF ACE AND Aβ
A few years after the first reported and somewhat confusing associations of ACE variation and AD risk, evidence that ACE might have a more direct role in AD pathology emerged and that may help with the interpretation of the reported ACE associations. Numerous in vitro and cell-culture based studies showed that ACE degraded Aβ [63–68]. There were conflicting conclusions regarding which amino acids in the Aβ peptide sequence that ACE cleaved; however, collectively the data provided evidence that ACE degraded Aβ at multiple locations [11]. These data that ACE could degrade Aβ provided another way of interpreting the emerging associations between ACE variants and AD, suggesting that the associations reflected varying heritability in ACE levels and thus peoples capacity to degrade Aβ, an important requirement in Aβ clearance that is thought to contribute to the development of AD [11].
In support of the in vitro studies, additional in vivo studies involving various murine chemically-induced or transgenic models of AD contributed valuable information. Early studies investigating the effect of acute and short-term ACE-inhibitor use on ACE-mediated degradation of Aβ in young mouse models showed no evidence of an effect on ‘steady-state’ levels of Aβ [69, 70]. However, studies involving older mice and longer use of the ACE-inhibitor captopril showed elevated Aβ deposition, as well as data supporting the role of ACE in the conversion of Aβ1-42 to Aβ1-40 in both mouse and human brain homogenates while also giving rise to other Aβ fragments [67, 68]. Not all studies agreed on the effect of ACE-inhibitors on Aβ pathology or other negative outcome measures of AD-like pathology in experimental models. For example, studies of the ACE-inhibitor perindopril given to mice [71] and rats [72], which had received intracerebroventricular (ICV)-injections of different Aβ species (ICV-Aβ), had better cognitive outcomes than untreated animals. Similarly, both cognitive function and cerebral blood flow improved in enalapril-treated streptozotocin (STZ)-treated diabetic rats; a rodent model proposed to simulate deficits in glucose and energy metabolism, and elevated oxidative stress, that are evident in AD [73]. Yet, enalapril also outperformed other ACE-inhibitors captopril, perindopril, and lisinopril at inhibiting the potentially protective mechanism of ACE-mediated conversion of Aβ1-42 to Aβ1-40 [67]. This finding was described as a possible explanation for why enalapril was found to be associated with increased incidence of AD in a population study [74]. In contrast, 2 months of captopril exposure did not alter Aβ pathology (measures of cognition were not measured) in the triple transgenic mouse model of AD [75], nor was there any cognitive benefit in ICV-Aβ injected mice given either of the ACE-inhibitors enalapril or imidapril [71]. Additional indirect evidence of potential ACE and other RAS involvement in AD, with links to Aβ pathology among other things, came in studies of angiotensin type 1 receptor (AT1R) antagonists (ARAs), that do not interfere with ACE function as do ACE-inhibitors but specifically inhibit ANGII signaling. In one study, the ARA candesartan improved cognitive function in STZ-treated mice [76], while losartan [77], valsartan, and telmisartan in transgenic mice [78] and in mice receiving ICV-Aβ [79] all had improved cognitive function and reduced Aβ pathology. The observations with both losartan and telmisartan are also worthy of note as both [79–81] are also thought, as well as some other ARAs, to be metabosartans [82] that also have agonistic properties on PPAR-γ (i.e., the peroxisome proliferator-activated receptor). PPAR-γ provides an additional interesting link between ARA function and AD because PPAR-γ activation has been implicated in the degradation and clearance of Aβ and decreases the activity of the Aβ promoting β-secretase BACE1 [83–85]. Indeed PPAR-γ has been proposed as a target for drug intervention in its own right, with drugs like rosiglitazone and pioglitazone being suggested as possible treatments for AD [86]. Yet, olmesartan, one of the ARAs that does not appear to have PPAR-γ agonist properties [85], also attenuated cerebrovascular dysfunction in the APP23 mouse model of AD and improved cognitive function in transgenic mice with continuous activation of the RAS [87]. Olmesartan also had a beneficial effect on cognitive function, independent of blood pressure effects, in other mice given ICV-Aβ [88]. However, a minority of data does not support these findings. Neither eprosartan nor valsartan affected AD-like pathology (where no cognitive data was obtained) in a triple transgenic mouse model of AD [75].
Another consideration in this apparent complex interaction between ACE and Aβ, aside from the interesting PPAR-γ side-story, lies in the structure of ACE itself and its two catalytic domains (see Fig. 1). Notably, the C-terminal domain of ACE is thought to be the primary domain through which ACE’s familiar role in ANGII formation is achieved, while the N-terminal domain is thought to be responsible for Aβ degradation as has been discussed [65, 89–91]. However, some studies suggested both domains were involved in Aβ degradation [92, 93] while additional findings suggest more intimate interactions between ACE and Aβ at the level of their expression. We have previously reported that ACE activity in cell culture increased following exposure of the cells to oligomeric forms of Aβ [61]. More recently our laboratory demonstrated that ICV-Aβ induced a progressive rise in blood pressure in Dahl salt-sensitive rats with pre-existing hypertension due to a high-salt diet. There was no change in blood pressure in similarly treated normotensive rats [94]. This study also suggested that intracerebral Aβ may exacerbate hypertension, through demonstrable modulation of autonomic activity, suggesting that the development of AD may sometimes be a physiological response to reduced cerebral perfusion due to midlife hypertension, thus complicating the accumulation of Aβ within the brain [94].
Fig.1
Human ACE-1 structure and domains specificity. Schematic representation of human ACE-1 domains structure. The two homologous domains (N-domain and C-domain) have a catalytic active zinc binding site (HExxH). The N-domain and most of C-domain are extracellular. Both domains are linked by a linker sequence (LR), while a transmembrane (TM) domain joins the C-domain with an intracellular C-terminus (CT) (adapted from [89]). The figure illustrates how N- and C-domains of ACE-1 differentially perform the reported roles of Aβ cleavage (N-domain) and more widely recognized conversion by angiotensin converting activity of angiotensin I to angiotensin II (C-domain).
![Human ACE-1 structure and domains specificity. Schematic representation of human ACE-1 domains structure. The two homologous domains (N-domain and C-domain) have a catalytic active zinc binding site (HExxH). The N-domain and most of C-domain are extracellular. Both domains are linked by a linker sequence (LR), while a transmembrane (TM) domain joins the C-domain with an intracellular C-terminus (CT) (adapted from [89]). The figure illustrates how N- and C-domains of ACE-1 differentially perform the reported roles of Aβ cleavage (N-domain) and more widely recognized conversion by angiotensin converting activity of angiotensin I to angiotensin II (C-domain).](https://content.iospress.com:443/media/jad/2018/62-3/jad-62-3-jad171119/jad-62-jad171119-g001.jpg)
The different roles of ACE catalytic domains on Aβ degradation, and potentially of Aβ on ACE levels of expression, may explain some of the inconsistencies observed in the various in vitro and in vivo studies undertaken thus far. Differences may also relate to some of the inconsistencies reported in vivo because of the variable affinities of different ACE-inhibitors used as tools in these studies, for each of the two ACE domains (see Table 1 for a summary). These reported differences in ACE catalytic domains among ACE-inhibitors likely contribute to the complex and sometimes unclear picture that has emerged over the years regarding the effect of ACE-inhibitors in various population studies, where cognitive decline and dementia risk have been investigated and is discussed further below.
Table 1
Reported specificities of ACE catalytic domains and some licensed and experimental ACE-1 inhibitors
| Inhibitor | N-domain specificity | C-domain specificity | N- &C- domain specificity | References |
| Captopril | ++ | NONE | + | [90, 91, 95, 96] |
| Lisinopril | NONE | ++ | + | [90, 91, 95, 96] |
| Lisinopril-tryptophan | NONE | + | NONE | [96–98] |
| Enalapril | ++ | NONE | + | [91, 95, 96] |
| Ramipril | NONE | NONE | + | [96] |
| *RXP407 | ++ | NONE | NONE | [95, 96, 99] |
| *RXP380 | NONE | ++ | NONE | [95, 96, 99] |
Experimental compounds are highlighted by *. the degree of affinity is denoted by the number of +’s while NONE corresponds to no evidence of binding.
EARLY EVIDENCE OF AD-ASSOCIATED RAS CHANGES IN THE CENTRAL NERVOUS SYSTEM
Prior to the ACE gene associations studies in AD, there were already a few small studies hinting at RAS changes in AD. Increased levels (although originally described as activity) of ACE the enzyme were seen in some regions of brain tissue homogenates from AD cases, that also correlated with Aβ senile plaque load, compared with control brain tissue [46]. ACE-inhibitor binding (as a measure of ACE levels) was increased in the temporal cortex of tissue from AD patients compared to controls [47]. In contrast, no significant differences were found, between AD cases and controls, in ACE activity measured in frontal cortex derived microvessels [100], or in homogenates taken from a variable number brain regions taken from AD patients [101].
Other studies have examined ACE in CSF where both reduced ACE levels [102, 103] but also no differences in ACE activity or levels [104, 105] were reported in AD. In efforts to characterize RAS in AD histologically, increased neuronal and perivascular ACE immunoreactivity was found in parietal cortex tissue from AD patients [106], while increased ANGII and ANGII receptor (AT1R, AT2R) binding and immunoreactivity have also been found in AD brain [103]. While further study of these important RAS receptors would be very informative and timely, efforts toward this are likely to be challenging since many of the commercial antibodies currently available have now been demonstrated to be less specific than was originally thought [107, 108] and thus bringing some previous findings into some doubt. In summary, there have been a small number of studies that provided limited, but nonetheless interesting, supportive data to the suggested role of RAS also found in genetic studies. The findings in many of these experimental contexts tended to be small or borderline, but so too were the sizes of many studies. The fact that numerous studies were providing similar or supportive signals that there were AD-associated changes in the RAS was sufficient for us to want to continue to pursue clearer answers to the tantalizing signals that were appearing. The undertaking of larger studies was necessary.
THE IMBALANCED RAS IN ALZHEIMER’S DISEASE
The brain has its own locally-acting (i.e., paracrine) renin-angiotensin-system (RAS) that functions independently of, but also likely interacts with, the systemic RAS [109]. The RAS has been extensively detailed over the years to capture the continually gathering information that paints a picture of a system of considerable complexity where over the last decade or, more receptors and peptide agonists have been found to have numerous and sometimes unexpected functions [110, 111]. Alternate regulatory pathways have also been identified that give rise some of various metabolites of the ANGII peptide, that is probably the single most biologically important peptide in the RAS (for detailed review see [112]).
In Fig. 2, the main elements of the RAS are presented and where for illustration purposes, some of the elements are compartmentalized to represent what is known as the ‘classical’ RAS pathway that the ‘regulatory’ RAS pathway attempts to continually counterbalance. The ‘classical’ RAS involves the conversion by the enzyme renin of angiotensinogen to angiotensin I (ANGI), that in turn is converted to the vasoconstrictor angiotensin II (ANGII) by ACE. Within the classical RAS a delicate balance is struck between, the activation of the angiotensin II type 1 receptor (AT1R) by ANGII, the primary signaling pathway in RAS that causes vasoconstriction, which can be countered (i.e., by vasodilatation) by ANGII-mediated activation of the angiotensin II type 2 receptor (AT2R) [113]. Numerous drugs have been developed to help reduce either the production (Renin-inhibitors and ACE-inhibitors) or signaling mediated by ANGII (angiotensin receptor antagonists, ARAs) on AT1R as treatments to reduce the vasoconstrictive effects of ANGII that can help with the management of hypertension (see also Fig. 2). Angiotensin II can also be converted by aminopeptidase A (APA) to angiotensin III (ANGIII), and in turn to angiotensin IV (ANGIV), whereby ANGIII and ANGIV can mediate similar vasoconstrictive effects to those by ANGII, since they also bind and activate AT1R [114–117].
Fig.2
Summary of the RAS system, including the compartmentalization of RAS to illustrate components that are part of the ‘Classical’ RAS and the ‘Regulatory’ RAS. The Classical RAS revolves around the production of the vasoconstrictor angiotensin II by angiotensin I-converting enzyme (ACE), and possibly angiotensin III and angiotensin IV by the sequential actions of aminopeptidases-A and -N on angiotensin II and angiotensin III respectively, and resultant signaling through the angiotensin II type I receptor (AT1R). Signaling through AT1R is thought to be the main signaling process in RAS that increases blood pressure (denoted by the heaviest weight arrows). In contrast, stimulation of the angiotensin II type 2 receptor (AT2R), by angiotensin II serves to counteract effects of AT1R. The sites of action of currently licensed drugs, usually used for the treatment of hypertension are also indicated where Renin inhibitors and ACE inhibitors work to reduce the formation of angiotensin II, whereas angiotensin receptor antagonists serve to inhibit the binding of angiotensin II to AT1R and instead promote vasodilatory inducing stimulation of AT2R by angiotensin II. The ‘Regulatory RAS’ has a similar role to that of AT2R in working to reduce blood pressure, however this is achieved by the activity of angiotensin II converting enzyme 2 (ACE2) on angiotensin II to produce angiotensin1-7 that can also bind AT2R or bind its own Mas receptor (MasR) to reduce blood pressure as indicated by the arrows. Notable but perhaps less well-known functions of the RAS are the effects, as illustrated by various peptides binding to the Insulin Regulated Aminopeptidase receptor (IRAP) (or angiotensin II type IV receptor (AT4R)), AT1R and MasR respectively on neuronal signaling pathways that can contribute to learning and memory.
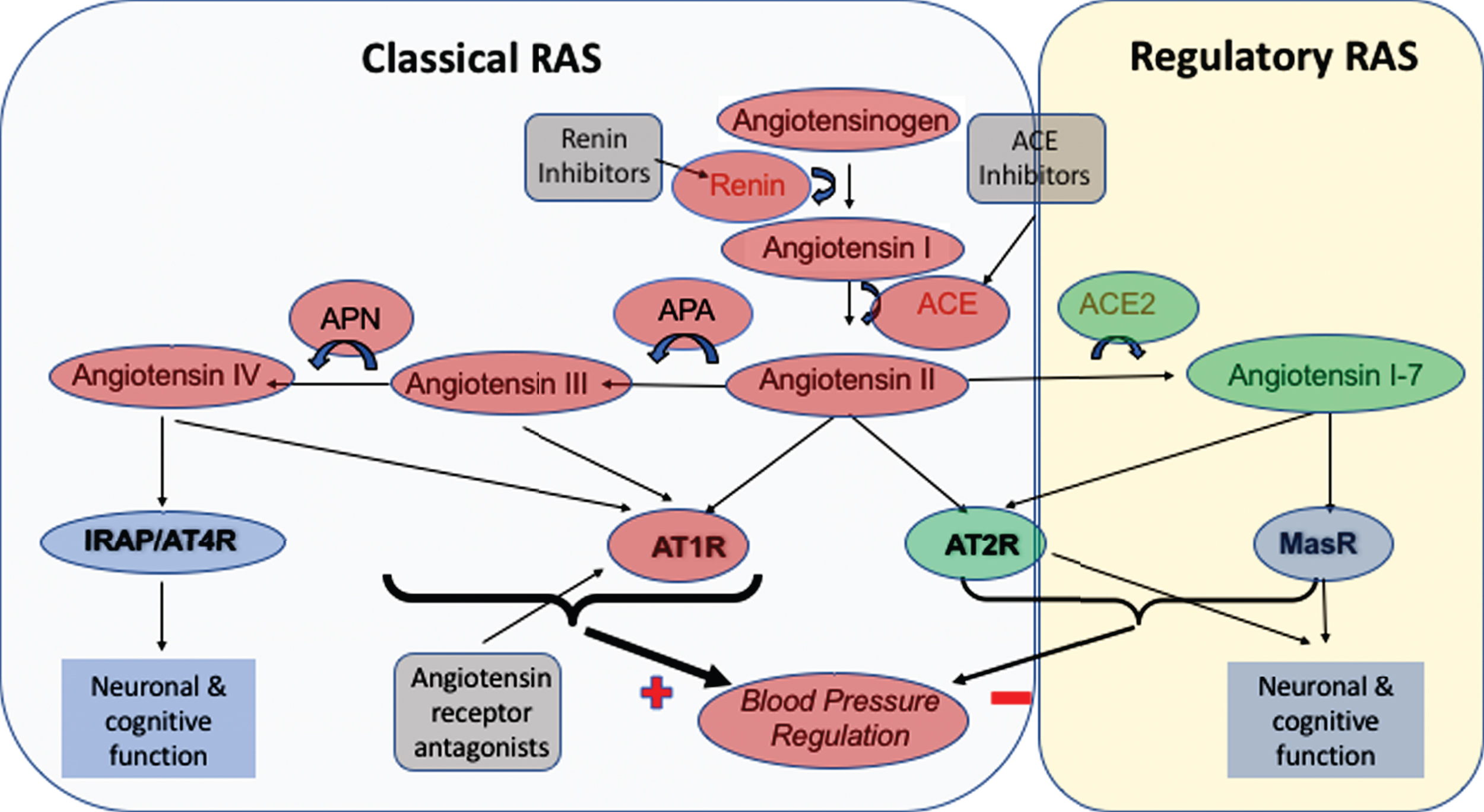
Collectively the pressor effects that result from ANGII, ANGIII, and perhaps ANGIV activation of AT1R are commonly considered to be the ‘classical’ actions of the RAS. The ‘regulatory’ pathway in RAS is somewhat newer and while it arguably shares the same stem components as the classical RAS that includes angiotensinogen, renin, and all the elements that contribute to the formation of ANGII, the main function of the regulatory RAS is the conversion by angiotensin converting enzyme 2 (ACE2) of ANGII to angiotensin 1–7 (ANG1-7). This peptide binds and activates the Mas receptor (MasR) to mediate a vasodilatory effect that counters the ‘pressor’ effects mediated through AT1R in the classical pathway [118]. Thus, the natural balance between the classical and regulatory RAS pathways is an inherent component of how blood pressure is normally regulated, and where other effects resulting from AT1R signaling (see below), are determined by the comparative activity of ACE relative to ACE2. Over the last decade our group has led a number of studies that investigated the RAS in postmortem tissue taken from people with AD and non-demented elderly to provide more data to inform the observations from various preclinical studies. Our first studies found increased ACE activity that was positively correlated with parenchymal Aβ load, as well as increased perivascular ACE immunoreactivity that was positively associated with the severity of CAA (i.e., Aβ deposition in blood vessels) [119]. We replicated these observations with additional measurements that took greater consideration of neuronal density, wherein ACE is normally abundant, that showed the AD-associated changes to ACE were even greater than previously shown because the ACE activity was higher despite significant neuronal loss that is typical in AD patients. Furthermore, we found that the elevated ACE activity, correlated positively with the severity of tau pathology [61]. These findings led to speculation that the altered ACE activity in AD was consequential to over production of ANGII where its multifunctional effects (see below) on various pathways contributed widely to the pathogenesis of AD. In view of the other pre-clinical data suggesting the role of ACE in the degradation of Aβ, the concurrent elevations of ACE in AD, were also seen to potentially have some beneficial effects toward reducing Aβ burden. However our data to this point, whereby ACE activity correlated positively with parenchymal load rather than negatively as might be expected if ACE was going to have an ameliorating effect on Aβ levels; combined with our other findings of how oligomeric forms of Aβ increased ACE activity [61] suggested otherwise and cast some doubt as to whether in vivo in humans ACE did degrade Aβ.
Nonetheless, given the data suggesting that the brain RAS, particularly the classical pathway, was overactive in human tissue, further supported by the aforementioned findings from various in vivo models of AD (and reviewed by [120]). However, the potential role of the RAS regulatory pathway, as a potential modifier of what was assumed to be elevated ANGII levels and signaling in AD had yet to be explored.
We recently showed that ACE2 activity was significantly reduced in AD in the same cohort of samples we had previously reported significant elevations in ACE [121]. The association between reduced ACE2 and AD also had stronger inverse correlations (than seen for ACE) with both parenchymal Aβ burden and tau pathology and reduced ACE2 was also more common in people whom were carriers of the APOE epsilon 4 and ACE I alleles, that have been reported as genetic risk factors for AD [121]. Collectively, these data, that can be summarized as a high ratio (as a proxy measure of classical RAS function) in the activity of ACE:ACE2 in AD patients compared to controls. Until the findings of reduced ACE2 in AD, the previous findings of elevated ACE in AD not appearing to have a significant effect on Aβ levels cast doubt as to whether ACE had a role in Aβ degradation. However, further studies have proposed that both ACE2 and ACE can perform a sequential degradation of Aβ, whereby ACE2 mediates the conversion of Aβ43 to Aβ42, which then allows ACE to further degrade Aβ42 to Aβ40 and some other smaller Aβ fragments [121]. Thus, it is possible that the capacity of significantly elevated levels of ACE in AD to reduce Aβ, at least levels of CAA where Aβ40 is the predominant Aβ species, is greatly limited by the reduced levels of ACE2 in the brain that are required for the first step in a sequential process [121]. Furthermore, our first empirical measurements of elevated levels of ANGII and reduced levels of its counterpart ANG1-7 in human brain tissue reinforced the predominance of ANGII and AT1R signaling in the classical RAS pathway (see Fig. 3) over that of the ACE2-ANG1-7-MasR regulatory pathway in AD [121].
Fig.3
Summary of the observed changes in the RAS system in postmortem AD brain tissue. Changes to various components of the RAS in AD means that the actions of the ‘Classical’ RAS, involving the production of angiotensins II angiotensin III by the sequential actions of angiotensin II converting enzyme (ACE), and aminopeptidases-A and -N on angiotensin I, that their subsequent activation of the angiotensin II type I receptor (AT1R) to raise blood pressure are largely preserved or elevated (denoted by the heaviest weight arrows and heavier borders of components). In contrast, the changes to preserve the classical RAS in AD do not seem extend to angiotensin IV where pressor signaling via AT1R is reduced but so is the capacity to stimulate neuronal signaling that is important to normal cognitive function. The sites where currently licensed drugs, that could be potentially used for the treatment of AD are also illustrated and noticeably present to sites within the classical pathway that is overactive in AD. Renin inhibitors, work to reduce the activity of the RAS pathway as a whole, whereas ACE inhibitors work to reduce the formation of angiotensin II. The angiotensin receptor antagonists in contrast serve to inhibit the binding of angiotensin II (and other angiotensins to AT1R) to promote vasodilation through the stimulation of AT2R by angiotensin II that is also thought to be involved in cognitive function. Alzheimer’s related changes also show a clear down regulation of the ‘Regulatory RAS’ (denoted by black coloured stars) where the scope to initiate (neuronal) signaling through both AT2R and MasR is reduced, with that likely loss of function that may explain some elements of cognitive decline and that all stem from observed significant reductions in angiotensin II converting enzyme 2 (ACE2) activity seen in AD, and significant elevations (as highlighted with stronger lines) of ANGII and ANGIII and their likely increased signaling through AT1R.
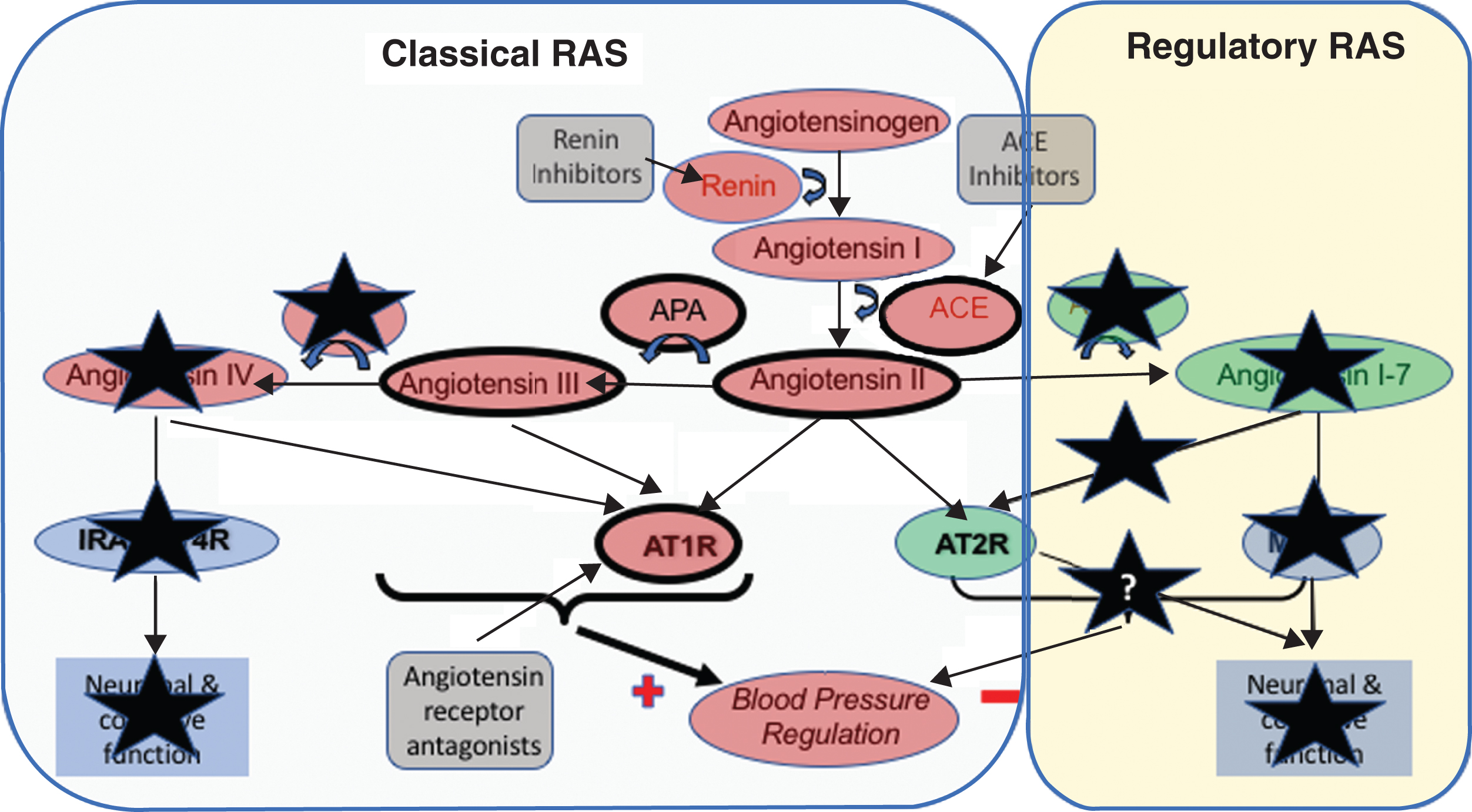
Noting that classical pathway is dominant in AD, it remained to be seen what happened downstream of ANGII formation. Angiotensin III (ANGIII), produced from ANGII by aminopeptidase A (APA), and with the ability to mediate similar pressor effects (Fig. 2) to ANGII via AT1R and AT2R [122, 123] warranted investigation [114]. We found that like ANGII, ANGIII levels were increased in postmortem AD brain tissue and similarly correlated strongly with parenchymal Aβ and tau load [124]. The increased ANGIII levels in AD reinforced the predominance of the classical pathway [121] resulting not only from reduced activity of the regulatory pathway, but also due to dysregulation of the APA/APN/ANGIV/IRAP(AT4R) elements of the pathway (Fig. 3). This was supported by an indication of increased APA activity, and significantly reduced APN activity, thereby maintaining higher levels of ANGIII (and classical pathway signaling) through reduced conversion to ANGIV [124]. Together these data suggest that ANGIII level, and the enzymes involved in its metabolism, may also contribute to the pathogenesis of AD.
There are additional considerations to make regarding these data. We are as yet unclear as why we found the discrepancies between APA level (that was significantly reduced) and APA activity (that was elevated, although not to a level of statistical significance). This may relate to post-translational modification of APA in AD that could change the activity of APA. We also showed that APA tended to localize within microglia surrounding Aβ plaques in AD, suggesting that certain pools of APA are recruited or produced in an immune response associated with AD pathology [124]. That the APA activity was the statistically no different between AD and controls, despite the concentration of the enzyme being significantly lower in the AD group, may reflect some compensatory changes to APA in response to the increased ANGII levels. What may also be of interest is the ability of APA, yet another RAS enzyme, to generate from Aβ42 a highly amyloidogenic and neurotoxic N-terminal truncated and pyroglutamated (AβpE3) Aβ42 species, that in itself could also contribute directly to AD pathogenesis [125, 126].
Our observed contrasting significant reduction in APN activity in AD was also supported by an apparent reduction of neuronal APN labeling in brain tissue sections, but our ELISA measurements of APN protein levels show no disease-associated differences. Importantly, reduced APN activity would reduce the levels of ANGIII conversion to ANGIV that could have important impact on downstream signaling pathways mediated by ANGIV through its receptor IRAP(AT4R) (Figs. 2 and 3) that has been shown to enhance learning and memory [127, 128].
The collective data over our series of studies on brain RAS points to excesses of ANGII and ANGIII (Fig. 3) that when considered alongside pre-clinical findings could increase AD pathology. In addition, as mentioned, the diminution of ACE2 in AD, that would reduce the effectiveness of ACE2:ACE1 mediated degradation of Aβ is also likely relevant. This has been supported in a large independent human postmortem series, where ARAs, that would inhibit the function of both ANGII and ANGIII, had less AD-related pathology compared with other hypertension treatment groups studied [129], and lower measures in CSF of measures of tau, but not Aβ, taken longitudinally [130]. The dysregulation of APN mediated production of ANGIV and loss of signaling that is important to memory is also relevant [127, 128], but becomes even more important when it is noted alongside data that ANG1-7 signaling via MasR also mediates long-term potentiation [131]. Thus the reductions in ACE2 and APN activity seen in AD, resulting in reduced formation of ANG1-7 and ANGIV respectively, may adversely impact on learning and memory processes (Fig. 3), as could the fact that high levels of ANGII and potentially ANGIII may inhibit ACh release [47, 132, 133].
TRANSLATING RAS STUDIES AT THE BENCH TO THE BEDSIDE FOR ALZHEIMER’S DISEASE
There is now a convincing body of data from numerous pre-clinical investigations that support how the RAS is altered and thus is involved in the pathogenesis of AD. The challenge is now to identify if reductions of the classical RAS, which is possible through drug re-purposing approaches, may have therapeutic potential in AD [25, 134]. Fortunately, the fact that there are numerous RAS-acting drugs to choose from with a lot prevailing safety data in different populations, provides significant opportunities for AD research that are not usually available when attempts to meet similar challenges are approached by drug development strategies [25]. Studies conducted over two decades have consistently shown that vascular factors increase the risk of dementia and AD. Hypertension in midlife [135, 136] and late life [137, 138]; diabetes mellitus [139, 140]; arterial stiffness [141]; atrial fibrillation [142]; and stroke [143] are but a few of the reported risk factors for AD. There have been conflicting conclusions [144, 145], although some of these likely relate to the methods and outcome measures studied (reviewed in [146]).
Detailed discussion of mechanisms proposed for reported associations between AD and some of these cardiovascular and metabolic syndrome factors is beyond the scope of this review, but detailed reviews are available for a number of these (hypertension [147], diabetes mellitus [148, 149], arterial stiffness [150], and stroke [151, 152]), where the importance of the RAS is discussed.
8.1Involvement of RAS in the incidence and progression of Alzheimer’s disease
Numerous clinical and population studies have, on the whole, provided evidence that RAS-acting drugs may outperform other anti-hypertensives in reducing the incidence of AD [74, 153–155] and the rate of progression of cognitive decline or conversion from milder forms of cognitive impairment to dementia [74, 156–163]. Similarly, there were supportive findings from meta-analyses [164, 165]; secondary investigations of dementia outcomes in hypertension trials, or measures of cognitive function in hypertensive patients taking RAS drugs [166–172]; or in AD trials of new interventions where cardiovascular medication history was also available [173–175]. Surprisingly, there have been few direct intervention trials of RAS-acting drugs in AD, and these were so small (n = 13 [176] and n = 30 [177]) that the conclusions that can be drawn are naturally limited. Other studies have described how centrally-acting RAS drugs may slow rates of conversion to dementia in African-Americans [178], or how the ARA candesartan outperformed lisinopril (ACE-inhibitor) and the calcium channel blocker hydrocholorothiazide [179] in executive function tests in a small, mainly Caucasian, population with mild cognitive impairment (MCI).
Nonetheless, there have been conflicting results from some studies (reviewed in [180]) based on some individual population studies [181–183] or meta-analyses [184, 185], where no overall benefits for lower rates of AD or reducing cognitive decline was observed. Yet, in some studies, the grouping of all RAS drugs together (i.e., combining ARAs and ACE-inhibitors) was undertaken. From data summarized here, the combining of all RAS drugs in this way, while defensible from the perspective of summarizing the collective inhibition of ANGII signaling, is overly simplistic with respect to AD, particularly so until the question as to the level to which ACE degrades Aβ is clarified.
8.2Additional roles of RAS in AD pathology: beyond blood pressure in AD
Data from many pre-clinical and clinical studies converge to support the potential involvement of the RAS in AD. Much of this involvement has focused on the role of RAS, not only in terms of potential relevance to blood pressure regulation and AD risk but also towards some elements of AD pathology as has been reported in a number of animal and human tissue studies. However, there remains a lack of clarity regarding whether early cerebrovascular disease is a fundamental precursor to the development of AD pathology [186]. The RAS may contribute to altered blood-brain barrier (BBB) permeability and cognition [187], while ANGII-induced hypertension worsened Aβ neuropathology in a transgenic mouse model of AD [188]. Similarly, ANGII administered centrally to non-transgenic rodents, by intracerebroventricular injection, stimulated Aβ production and tau-phosphorylation [189]. In contrast, an alternative question that is relevant is whether cardiovascular changes are secondary to the development of AD pathology, as has been proposed in some population studies [144]. Reports that Aβ increased ACE activity in cultured neuroblastoma cells [61]; where Aβ40 exacerbated pre-existing hypertension in rodents [94]; and where Aβ-mediated neurovascular uncoupling gave rise to the reactive oxygen species and oxidative stress that is associated with AD [190, 191] all support this possibility.
A primary or secondary role for hypertension in AD is conceivable, particularly so given the often lengthy and insidious time course in the evolution of AD [26] that coincides with the emergence and rising prevalence of hypertension in populations [192, 193]. Regardless, early cerebrovascular disturbances are central to the concept of the vascular hypothesis of AD (reviewed in [194]), where it also may serve as a likely determinant of the additional development of Aβ in blood vessels of the brain, which is very common in AD [8]. Thus, intervention will likely improve any of a number of possible aspects of the pathogenesis of AD, and the RAS, based on evidence presented, is now a credible target to try and achieve this. Yet, this story has more to offer. There is also other evidence that warrants some mention, that demonstrates an even wider involvement of RAS in AD, and in doing so further emphasizes the candidacy of RAS as a pathway in which intervention could achieve some positive outcomes clinically, socially and economically.
The overactive classical RAS pathway present in AD (Fig. 3) could cause ANGII-mediated inhibition of ACh release, as reported in various animal and human brain tissue studies [47, 132, 133]. More recent pre-clinical studies where deficits in spatial and short-term memory mechanisms and pathological processes that require cholinergic involvement were ameliorated by RAS acting agents [195, 196]. Thus, the targeting of ANGII could not only benefit pathological mechanisms in AD mediated by Aβ and tau, but also potentially enhance cholinergic release and signaling. RAS-acting drugs may thus potentially supplement existing anti-cholinesterase treatment strategies in AD. Recent findings that other receptors in RAS, namely IRAP(AT4R) that can respectively enhance learning and memory [127, 128], and MasR that mediates long-term potentiation [131], illustrates not only that AT1R signaling may be detrimental in AD, but also that the loss of activity of these other receptors is significant and presents the opportunity for further targets for intervention.
As mentioned, an inflammatory hypothesis has also been proposed for AD [20]. This has been the focus of a number of clinical trials in AD where the pro-inflammatory mediator TNFα has been a major focus [197]. Notably, the actions of some inflammatory mediators may be downstream effects of RAS over-activation since ANGII mediates pro- and anti-inflammatory effects, that are very prominent in AD, by activating TNFα and TGFβ signaling pathways, respectively [198, 199]. ANGII also contributes to BBB maintenance [200]; to cell survival via the interplay of AT1R and AT2R receptor signaling [198]; and to calcium signaling that is also relevant to the pathogenesis of AD [201–203]. Thus, there are a number of other important processes that are all additionally relevant to the pathogenesis of AD, and where an overactive RAS could contribute to, clearly reinforcing the case for RAS blockers to be considered as possible interventions for AD.
UNRESOLVED ISSUES IN THE ANGIOTENSIN HYPOTHESIS OF ALZHEIMER’S DISEASE: FUTURE RESEARCH NEEDS
There are a number of important unresolved issues that warrant further investigation.
(A) Does ACE degrade Aβ?
One of the most important issues to clarify is whether ACE degrades Aβ and if so, what might be the consequences of current widespread prescription of ACE-inhibitors as a frontline treatment for hypertension. There is already evidence that only the N-domain catalytic site on ACE is responsible for Aβ cleavage, however, there are also conflicting reports of the extent to which different ACE-inhibitors bind to the ACE catalytic domains (Table 1). Until this question is resolved, involving more systematic study of ACE-inhibitors in relation to AD pathology, it is possible that a subset of ACE-inhibitors, while acting to reduce blood pressure in people with hypertension, represent modifiable risk factors (i.e., potentially avoidable) for the progressive accumulation of Aβ that can give rise to CAA and AD. Such investigations could help to identify the ACE inhibitors that specifically target the C-domain catalytic site on ACE and so can continue to serve as effective treatments for hypertension that millions of people worldwide require and benefit from. In addition, it can also potentially provide some contribution towards the primary prevention of AD, since potential interference with natural Aβ-degrading mechanisms could be avoided.
By tackling this issue, it will also have some bearing on the related question of where in the Aβ peptide sequence that ACE cleavage occurs. The different locations reported to date is likely the result of different experimental approaches used (discussed in more detail in [204]) to try and determine this [63–66, 68, 92]. Clearer understanding of what, if any, subsets of ACE-inhibitors may afford some risk in AD, could also clarify which ACE-inhibitors could serve as ‘tool drugs’ in experiments to better characterize the locations and dynamics of ACE mediated degradation of Aβ.
Post-translational modifications, and in rare cases, autosomal dominant inheritance of genetic mutations in the amyloid precursor protein (APP) gene that cause a very small proportion of AD cases, coincide with or are nearby to sites on Aβ that have been reported to be the sites of ACE-mediated cleavage of Aβ [66, 205, 206]. Some modifications, such as the isomerization of Aspartate-7 (Asp-7) residue, that occurs increasingly in aging [207], and which has been found in Aβ senile plaques, may determine the levels of insolubility and oligomerization of Aβ fragments and thus the resistance of Aβ to enzymatic cleavage [66, 207–209].
A more in-depth knowledge of the nature of post-translational modifications of Aβ and the impact of these on the affinity of ACE for Aβ would provide helpful clarifications on whether such modifications have any bearing on whether ACE-inhibitors interfere with Aβ cleavage and clearance mechanisms. In other words, do such modifications prevent ACE-mediated cleavage of Aβ, and thus the concerns about ACE-inhibitors become irrelevant. Unfortunately, such modifications may not be able to come to the rescue of ACE-inhibitors as there is already supportive evidence in populations where ACE-inhibitors were associated with increased hazard ratios for incidence of AD [74] and mortality [210, 211] that need to be continually borne in mind and further studied.
(B) How important is the blood-brain barrier in relation to RAS blocking drugs?
There are conflicting findings regarding the effect of RAS-acting drugs in AD, whether they cross the BBB or not and thus ACE-inhibitors cannot be considered as interchangeable with respect to AD [212, 213]. Such concerns apply more to ACE-inhibitors than ARAs, since there is less ambiguity regarding the latter and their abilities to cross the BBB [214]. As discussed, there are supportive findings that centrally acting ACE-inhibitors (i.e., those that cross the BBB) had less cognitive decline than people taking peripherally acting ACE-inhibitors [74, 157]. Another recent study from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) supports this whereby BBB penetrating ACE-inhibitors and ARAs had superior memory performance and less white matter hyperintensities volume [215]. There have also been reports that the cognitive decline of users of peripherally acting ACE-inhibitors declined more rapidly and had a higher hazard ratio for AD incidence than people taking the centrally active ACE-inhibitors [74]. We found evidence that levels of ACE, while having a beneficial effect on lowering Aβ levels, may also be associated with greater vascular pathology in AD patients [216, 217]. These observations reinforce the need to clarify the true nature of ACE-inhibitors and ACE catalytic domains and the potential it would bring to not only reduce ANGII formation, but also avoid interfering with ACE-mediated cleavage of Aβ. Other studies have showed variable protective benefits between ACE-inhibitors and ARAs in relation to the incidence of AD and dementia [154, 155, 210, 218], and usually ARAs are superior. The possible explanation for this being that they exclusive inhibit ANGII (and ANGIII) signaling and do not interfere with ACE activity that affords some Aβ-lowering benefit.
There is a persuasive argument that ACE-inhibitors should not be considered as interchangeable in relation to risk of AD and in people with AD needing medication to treat hypertension [212]. The fact that there are conflicting reports as to the level of BBB penetration of a number of ACE-inhibitors does not help either [219–223]. There have been efforts to better understand the BBB penetrability of these compounds following oral administration [221, 224–230]; however, the majority of these studies were in experimental conditions targeted to inform hypertension research, rather than AD research, where BBB integrity and progressive failure is perhaps more marked as part of AD pathogenesis [188]. In short, systematic re-examination of (at least) the more commonly used ACE-inhibitors, to determine those unlikely to interfere with ACE-mediated degradation of Aβ, is now imperative. This will not least help to prioritize what ACE-inhibitors that may be amenable for future study in AD as interventions, but also potentially inform revisions to current guidelines regarding prescribing approaches in the management of hypertension.
(C) To what extent is cognitive function influenced by RAS signaling?
There is already evidence that ANGII (and potentially ANGIII) signaling through AT1R has an anti-cholinergic effect. The tantalizing data of ANGIV and ANG1-7 mediated effects on learning and memory and long term potentiation warrant greater study, not only in AD but also in general age-associated cognitive decline [127, 128, 131, 231] for review [232, 233]). There is also significant scope for greater understanding of the mechanisms by which AT2R activation, that may result from ARAs [234], may contribute to some of the observed protective functions discussed in this review and by others where AT2R has numerous relevant functions in neurons, including modulation of neuronal excitability and its activation of PPAR that has already been described as important in AD pathology (reviewed in [235]).
THE ULTIMATE TEST: CLINICAL TRIALS OF RAS BLOCKADE IN ALZHEIMER’S DISEASE
The convergence of numerous lines of supportive evidence has now positioned the RAS as a credible target for intervention in AD, which is sorely needed to increase the currently limited therapeutic options available for AD [18]. The ultimate test will be that by clinical trial and fortunately, thanks to the readily availability of RAS acting drugs, a number of trials of varying sizes, have now commenced to explore various questions regarding the role of RAS in the development and pathology AD.
The first such trial to commence and likely first to finish is the UK-based (with a recruitment target of n = 228) Phase II multi-center RADAR trial of losartan compared to placebo in hypertensive and normotensive AD patients (Study ISRCTN93682878 at http://www.isrctn.com/ISRCTN93682878) where the primary outcome is change to MRI-based measures of brain structure and volume after 12 months of treatment [32]. A similar design and sized (SARTAN-AD) Phase II trial in hypertensive AD patients will compare perindopril with telmisartan (Study NCT02085265 at https://clinicaltrials.gov/ct2/show/NCT02085265). The smaller pilot Phase I (n = 66) HEART study (Study NCT02471833 at https://clinicaltrials.gov/ct2/show/NCT02471833) will compared two doses of telmisartan against placebo for effects on CSF levels of RAS components in African Americans at increased risk of AD [236]. A similarly sized (n = 72) CEDAR study (Study NCT02646982 at https://clinicaltrials.gov/ct2/show/NCT02646982) will compare the effect of candesartan and placebo on a number of cardiovascular outcome measures in people with MCI, while the CALIBREX study (Study NCT01984164 at https://clinicaltrials.gov/ct2/show/NCT01984164) will compare lisinopril with candesartan for effects on the primary outcome of executive function in people with hypertension and MCI. Finally, the rrAD study (Study NCT02913664 at https://clinicaltrials.gov/ct2/show/NCT02913664) will compare the effects of losartan and amlodipine in conjunction with aerobic exercise training on cognitive performance in older adults who have high risk for AD.
While none of these trials are sufficiently large to provide definitive proof of RAS involvement in AD, and instead are designed to inform larger Phase III studies, they serve as the first formal gold-standard tests of RAS as a target for intervention in AD patients and also elderly with MCI. Thus, the findings of this new collection of important studies are eagerly awaited, not only to improve our understanding of RAS involvement in AD, but also to provide insights into the vital lessons that can be learned to enhance the study design of any future definitive trials. These would aspire to be as inclusive as possible for participants (hypertensive and normotensive), and as naturalistic as possible in terms of fitting well with standard care, as well as informing what might be the optimal diagnostic groups (e.g., AD or MCI) to include.
CONCLUSIONS
This review has attempted to describe what has been the maturation of the evidence that implicates the RAS in AD and gives credence to the angiotensin hypothesis for AD. Converging evidence from numerous pre-clinical and clinical lines of research into the RAS in AD may finally explain widely reported, less well understood, associations between hypertension and AD. This is also compatible and consistent with the vascular hypothesis of AD that continues to gain support. In the last two decades, the angiotensin hypothesis has come of age from relatively spasmodic and unrelated lines of research enquiry towards more focused and sometimes increasingly larger or more rigorous studies, the findings of which have now provided sufficient evidence to justify the clinical trials that are now underway. There remain unresolved issues that warrant further and careful research but which have the potential to be impactful on a global scale in their own right. How certain hypertension treatments might require removal from normal use, and in doing so help focus in on those that have the best long-term benefits against both hypertension and the development of AD is a key example. As a researcher of RAS in AD for nearly two decades, these are genuinely exciting times with the results of ongoing clinical trials keenly awaited. The results of these trials will hopefully provide some positive results to pave the way for future Phase III trials that can exploit the plethora of readily available generic drugs, many with extensive safety data and most because they exist in generic form, will be highly economical options for publicly funded health care systems, where they can be made widely available to all patients in need.
ACKNOWLEDGMENTS
PGK’s research on various aspects of the renin angiotensin system involvement in AD would not have been possible without the support of a long-term Fellowship from the Sigmund Gestetner Foundation. Individual projects over the years would not have been possible without the support of Bristol Research into Care of the Elderly (BRACE), NIHR-EME, Alzheimer’s Research UK, the Perros Trust, the Bright Focus Foundation, University of Bristol Alumni Foundation and North Bristol NHS Trust. The author would also like to thank Dr. Noura Al-Mulhim for some assistance in the generation of Fig. 1 and Table 1.
The author would also humbly like to thank the collective efforts of all of the researchers who have worked tirelessly over the last two decades or more to generate the valuable findings and insights that the wider dementia community can use to increase our understanding not only of Alzheimer’s disease but also more widely of the renin angiotensin system in the brain. These efforts have without question progressed this area of research enquiry from relative obscurity, mostly at the laboratory bench, to a very relevant, expanding and credible line of research into Alzheimer’s disease and other forms of age-related cognitive impairment. There is much hope generated by research to date and now the next important steps will lie with the generosity and altruism of people living with cognitive impairment and dementia, who will be our much-needed researcher partners, in the vital future clinical studies that will truly test the veracity of the hypotheses we have all been generating, and hopefully provide meaningful breakthroughs that we can collectively be proud to have contributed to.
The author’s disclosure is available online (https://www.j-alz.com/manuscript-disclosures/17-1119r1).
REFERENCES
[1] | Wu YT , Fratiglioni L , Matthews FE , Lobo A , Breteler MM , Skoog I , Brayne C ((2016) ) Dementia in western Europe: epidemiological evidence and implications for policy making. Lancet Neurol 15: , 116–124. |
[2] | Lemolo F , Duro G , Rizzo C , Castiglia L , Hachinski V , Caruso C ((2009) ) Pathophysiology of vascular dementia. Immun Ageing 6: , 13. |
[3] | Schneider JA , Arvanitakis Z , Leurgans SE , Bennett DA ((2009) ) The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 66: , 200–208. |
[4] | Love S ((2004) ) Contribution of cerebral amyloid angiopathy to Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75: , 1–4. |
[5] | Love S , Miners JS ((2016) ) Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol 131: , 645–658. |
[6] | Love S , Miners JS ((2017) ) Small vessel disease, neurovascular regulation and cognitive impairment: Post-mortem studies reveal a complex relationship, still poorly understood. Clin Sci (Lond) 131: , 1579–1589. |
[7] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[8] | Weller RO , Subash M , Preston SD , Mazanti I , Carare RO ((2008) ) Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol 18: , 253–266. |
[9] | Deane R , Sagare A , Zlokovic BV ((2008) ) The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr Pharm Des 14: , 1601–1605. |
[10] | Miners JS , Baig S , Palmer J , Palmer LE , Kehoe PG , Love S ((2008) ) Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol 18: , 240–252. |
[11] | Miners JS , Barua N , Kehoe PG , Gill S , Love S ((2011) ) Abeta-degrading enzymes: Potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol 70: , 944–959. |
[12] | Marciani DJ ((2017) ) Rejecting the Alzheimer’s disease vaccine development for the wrong reasons. Drug Discov Today 22: , 609–614. |
[13] | Hung SY , Fu WM ((2017) ) Drug candidates in clinical trials for Alzheimer’s disease. J Biomed Sci 24: , 47. |
[14] | Bartus RT , Dean 3rd RL , Beer B , Lippa AS ((1982) ) The cholinergic hypothesis of geriatric memory dysfunction. Science 217: , 408–414. |
[15] | Pakaski M , Kalman J ((2008) ) Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochem Int 53: , 103–111. |
[16] | Davies P , Maloney AJ ((1976) ) Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2: , 1403. |
[17] | Whitehouse PJ , Price DL , Struble RG , Clark AW , Coyle JT , Delon MR ((1982) ) Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 215: , 1237–1239. |
[18] | Sindi S , Mangialasche F , Kivipelto M ((2015) ) Advances in the prevention of Alzheimer’s disease. F1000Prime Rep 7: , 50. |
[19] | de la Torre JC ((2010) ) The vascular hypothesis of Alzheimer’s disease: Bench to bedside and beyond. Neurodegener Dis 7: , 116–121. |
[20] | McGeer PL , McGeer EG ((2013) ) The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol 126: , 479–497. |
[21] | Swerdlow RH , Burns JM , Khan SM ((2014) ) The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim Biophys Acta 1842: , 1219–1231. |
[22] | Padurariu M , Ciobica A , Lefter R , Serban IL , Stefanescu C , Chirita R ((2013) ) The oxidative stress hypothesis in Alzheimer’s disease. Psychiatr Danub 25: , 401–409. |
[23] | Morris GP , Clark IA , Vissel B ((2014) ) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun 2: , 135. |
[24] | Shelanski M , Shin W , Aubry S , Sims P , Alvarez MJ , Califano A ((2015) ) A systems approach to drug discovery in Alzheimer’s disease. Neurotherapeutics 12: , 126–131. |
[25] | Corbett A , Pickett J , Burns A , Corcoran J , Dunnett SB , Edison P , Hagan JJ , Holmes C , Jones E , Katona C , Kearns I , Kehoe P , Mudher A , Passmore A , Shepherd N , Walsh F , Ballard C ((2012) ) Drug repositioning for Alzheimer’s disease. Nat Rev Drug Discov 11: , 833–846. |
[26] | Selkoe DJ ((2011) ) Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med 17: , 1060–1065. |
[27] | Chaudhury S , Patel T , Barber IS , Guetta-Baranes T , Brookes KJ , Chappell S , Turton J , Guerreiro R , Bras J , Hernandez D , Singleton A , Hardy J , Mann D , ARUKConsortium, Morgan K ((2018) ) Polygenic risk score in postmortem diagnosed sporadic early-onset Alzheimer’s disease. Neurobiol Aging 62: , 244.e1–244.e8. |
[28] | Escott-Price V , Myers AJ , Huentelman M , Hardy J ((2017) ) Polygenic risk score analysis of pathologically confirmed Alzheimer disease. Ann Neurol 82: , 311–314. |
[29] | Escott-Price V , Shoai M , Pither R , Williams J , Hardy J ((2017) ) Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging 49: , 214e217–214e211. |
[30] | Roubroeks JAY , Smith RG , van den Hove DLA , Lunnon K ((2017) ) Epigenetics and DNA methylomic profiling in Alzheimer’s disease and other neurodegenerative diseases. J Neurochem 143: , 158–170. |
[31] | Ferrante M , Conti GO ((2017) ) Environment and neurodegenerative diseases: An update on miRNA role. Microrna 6: , 157–165. |
[32] | Kehoe PG , Blair PS , Howden B , Thomas DL , Malone IB , Horwood J , Clement C , Selman LE , Baber H , Lane A , Coulthard E , Passmore AP , Fox NC , Wilkinson IB , Ben-Shlomo Y ((2018) ) The rationale and design of the reducing pathology in Alzheimer’s disease through Angiotensin TaRgeting (RADAR) Trial. J Alzheimers Dis 61: , 803–814. |
[33] | Soubrier F , Cambien F ((1994) ) The angiotensin I-converting enzyme gene polymorphism: Implication in hypertension and myocardial infarction. Curr Opin Nephrol Hypertens 3: , 25–29. |
[34] | Kehoe PG , Russ C , McIlory S , Williams H , Holmans P , Holmes C , Liolitsa D , Vahidassr D , Powell J , McGleenon B , Liddell M , Plomin R , Dynan K , Williams N , Neal J , Cairns NJ , Wilcock G , Passmore P , Lovestone S , Williams J , Owen MJ ((1999) ) Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat Genet 21: , 71–72. |
[35] | Chapman J , Wang N , Treves TA , Korczyn AD , Bornstein NM ((1998) ) ACE, MTHFR, factor V Leiden, and APOE polymorphisms in patients with vascular and Alzheimer’s dementia. Stroke 29: , 1401–1404. |
[36] | Scacchi R , De Bernardini L , Mantuano E , Vilardo T , Donini LM , Ruggeri M , Gemma AT , Pascone R , Corbo RM ((1998) ) DNA polymorphisms of apolipoprotein B and angiotensin I-converting enzyme genes and relationships with lipid levels in Italian patients with vascular dementia or Alzheimer’s disease. Dement Geriatr Cogn Disord 9: , 186–190. |
[37] | Bertram L , McQueen MB , Mullin K , Blacker D , Tanzi RE ((2007) ) Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat Genet 39: , 17–23. |
[38] | Rigat B , Hubert C , Alhenc-Gelas F , Cambien F , Corvol P , Soubrier F ((1990) ) An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 86: , 1343–1346. |
[39] | Danilov S , Savoie F , Lenoir B , Jeunemaitre X , Azizi M , Tarnow L , Alhenc-Gelas F ((1996) ) Development of enzyme-linked immunoassays for human angiotensin I converting enzyme suitable for large-scale studies. J Hypertens 14: , 719–727. |
[40] | Cox R , Bouzekri N , Martin S , Southam L , Hugill A , Golamaully M , Cooper R , Adeyemo A , Soubrier F , Ward R , Lathrop G , Matsuda F , Farrall M ((2002) ) Angiotensin-1-converting enzyme (ACE) plasma concentration is influenced by multiple ACE-linked quantitative trait nucleotides. Hum Mol Genet 11: , 2969–2977. |
[41] | McKenzie CA , Sinsheimer JS , Adeyemo AA , Cox RD , Southam L , Hugill A , Bouzekri N , Lathrop M , Forrester TE , Cooper RS , Ward R ((2005) ) SNP haplotypes in the angiotensin I-converting enzyme (ACE) gene: Analysis of Nigerian family data using gamete competition models. Ann Hum Genet 69: , 227–232. |
[42] | Villard E , Tiret L , Visvikis S , Rakotovao R , Cambien F , Soubrier F ((1996) ) Identification of new polymorphisms of the angiotensin I-converting enzyme (ACE) gene, and study of their relationship to plasma ACE levels by two-QTL segregation-linkage analysis. Am J Hum Genet 58: , 1268–1278. |
[43] | Soubrier F , Martin S , Alonso A , Visvikis S , Tiret L , Matsuda F , Lathrop G , Farrall M ((2002) ) High-resolution genetic mapping of the ACE-linked QTL influencing circulating ACE activity. Eur J Hum Genet 10: , 553–561. |
[44] | Binevski PV , Sizova EA , Pozdnev VF , Kost OA ((2003) ) Evidence for the negative cooperativity of the two active sites within bovine somatic angiotensin-converting enzyme. FEBS Lett 550: , 84–88. |
[45] | van Esch JH , van Gool JM , de Bruin RJ , Payne JR , Montgomery HE , Hectors M , Deinum J , Dive V , Jan Danser AH ((2008) ) Different contributions of the angiotensin-converting enzyme C-domain and N-domain in subjects with the angiotensin-converting enzyme II and DD genotype. J Hypertens 26: , 706–713. |
[46] | Arregui A , Perry EK , Rossor M , Tomlinson BE ((1982) ) Angiotensin converting enzyme in Alzheimer’s disease increased activity in caudate nucleus and cortical areas. J Neurochem 38: , 1490–1492. |
[47] | Barnes NM , Cheng CH , Costall B , Naylor RJ , Williams TJ , Wischik CM ((1991) ) Angiotensin converting enzyme density is increased in temporal cortex from patients with Alzheimer’s disease. Eur J Pharmacol 200: , 289–292. |
[48] | Elkins JS , Douglas VC , Johnston SC ((2004) ) Alzheimer disease risk and genetic variation in ACE: A meta-analysis. Neurology 62: , 363–368. |
[49] | Kehoe PG , Katzov H , Feuk L , Bennet AM , Johansson B , Wiman B , de Faire U , Cairns NJ , Wilcock GK , Brookes AJ , Blennow K , Prince JA ((2003) ) Haplotypes extending across ACE are associated with Alzheimer’s disease. Hum Mol Genet 12: , 859–867. |
[50] | Lehmann DJ , Cortina-Borja M , Warden DR , Smith AD , Sleegers K , Prince JA , van Duijn CM , Kehoe PG ((2005) ) Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer’s disease. Am J Epidemiol 162: , 305–317. |
[51] | Narain Y , Yip A , Murphy T , Brayne C , Easton D , Evans JG , Xuereb J , Cairns N , Esiri MM , Furlong RA , Rubinsztein DC ((2000) ) The ACE gene and Alzheimer’s disease susceptibility. J Med Genet 37: , 695–697. |
[52] | Meng Y , Baldwin CT , Bowirrat A , Waraska K , Inzelberg R , Friedland RP , Farrer LA ((2006) ) Association of polymorphisms in the Angiotensin-converting enzyme gene with Alzheimer disease in an Israeli Arab community. Am J Hum Genet 78: , 871–877. |
[53] | Kehoe PG , Katzov H , Andreasen N , Gatz M , Wilcock GK , Cairns NJ , Palmgren J , de Faire U , Brookes AJ , Pedersen NL , Blennow K , Prince JA ((2004) ) Common variants of ACE contribute to variable age-at-onset of Alzheimer’s disease. Hum Genet 114: , 478–483. |
[54] | Sleegers K , den Heijer T , van Dijk EJ , Hofman A , Bertoli-Avella AM , Koudstaal PJ , Breteler MM , van Duijn CM ((2005) ) ACE gene is associated with Alzheimer’s disease and atrophy of hippocampus and amygdala. Neurobiol Aging 26: , 1153–1159. |
[55] | Coon KD , Myers AJ , Craig DW , Webster JA , Pearson JV , Lince DH , Zismann VL , Beach TG , Leung D , Bryden L , Halperin RF , Marlowe L , Kaleem M , Walker DG , Ravid R , Heward CB , Rogers J , Papassotiropoulos A , Reiman EM , Hardy J , Stephan DA ((2007) ) A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 68: , 613–618. |
[56] | Li H , Wetten S , Li L , St Jean PL , Upmanyu R , Surh L , Hosford D , Barnes MR , Briley JD , Borrie M , Coletta N , Delisle R , Dhalla D , Ehm MG , Feldman HH , Fornazzari L , Gauthier S , Goodgame N , Guzman D , Hammond S , Hollingworth P , Hsiung GY , Johnson J , Kelly DD , Keren R , Kertesz A , King KS , Lovestone S , Loy-English I , Matthews PM , Owen MJ , Plumpton M , Pryse-Phillips W , Prinjha RK , Richardson JC , Saunders A , Slater AJ , St George-Hyslop PH , Stinnett SW , Swartz JE , Taylor RL , Wherrett J , Williams J , Yarnall DP , Gibson RA , Irizarry MC , Middleton LT , Roses AD ((2008) ) Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol 65: , 45–53. |
[57] | Thornton-Wells TA , Moore JH , Martin ER , Pericak-Vance MA , Haines JL ((2008) ) Confronting complexity in late-onset Alzheimer disease: Application of two-stage analysis approach addressing heterogeneity and epistasis. Genet Epidemiol 32: , 187–203. |
[58] | Lee JH , Cheng R , Graff-Radford N , Foroud T , Mayeux R ((2008) ) Analyses of the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study: Implication of additional loci. Arch Neurol 65: , 1518–1526. |
[59] | Schjeide BM , McQueen MB , Mullin K , Divito J , Hogan MF , Parkinson M , Hooli B , Lange C , Blacker D , Tanzi RE , Bertram L ((2009) ) Assessment of Alzheimer’s disease case-control associations using family-based methods. Neurogenetics 10: , 19–25. |
[60] | Kauwe J , Wang J , Mayo K , Morris J , Fagan A , Holtzman D , Goate A ((2009) ) Alzheimer’s disease risk variants show association with cerebrospinal fluid amyloid beta. Neurogenetics 10: , 13–17. |
[61] | Miners S , Ashby E , Baig S , Harrison R , Tayler H , Speedy E , Prince JA , Love S , Kehoe PG ((2009) ) Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: Differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res 1: , 163–177. |
[62] | Lambert JC , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , DeStafano AL , Bis JC , Beecham GW , Grenier-Boley B , Russo G , Thorton-Wells TA , Jones N , Smith AV , Chouraki V , Thomas C , Ikram MA , Zelenika D , Vardarajan BN , Kamatani Y , Lin CF , Gerrish A , Schmidt H , Kunkle B , Dunstan ML , Ruiz A , Bihoreau MT , Choi SH , Reitz C , Pasquier F , Cruchaga C , Craig D , Amin N , Berr C , Lopez OL , De Jager PL , Deramecourt V , Johnston JA , Evans D , Lovestone S , Letenneur L , Morón FJ , Rubinsztein DC , Eiriksdottir G , Sleegers K , Goate AM , Fiévet N , Huentelman MW , Gill M , Brown K , Kamboh MI , Keller L , Barberger-Gateau P , McGuiness B , Larson EB , Green R , Myers AJ , Dufouil C , Todd S , Wallon D , Love S , Rogaeva E , Gallacher J , St George-Hyslop P , Clarimon J , Lleo A , Bayer A , Tsuang DW , Yu L , Tsolaki M , Bossù P , Spalletta G , Proitsi P , Collinge J , Sorbi S , Sanchez-Garcia F , Fox NC , Hardy J , Deniz Naranjo MC , Bosco P , Clarke R , Brayne C , Galimberti D , Mancuso M , Matthews F , European Alzheimer’s Disease Initiative (EADI), Genetic and Environmental Risk in Alzheimer’s Disease, Alzheimer’s Disease Genetic Consortium, Cohorts for Heart and Aging Research in Genomic Epidemiology, Moebus S , Mecocci P , Del Zompo M , Maier W , Hampel H , Pilotto A , Bullido M , Panza F , Caffarra P , Nacmias B , Gilbert JR , Mayhaus M , Lannefelt L , Hakonarson H , Pichler S , Carrasquillo MM , Ingelsson M , Beekly D , Alvarez V , Zou F , Valladares O , Younkin SG , Coto E , Hamilton-Nelson KL , Gu W , Razquin C , Pastor P , Mateo I , Owen MJ , Faber KM , Jonsson PV , Combarros O , O’Donovan MC , Cantwell LB , Soininen H , Blacker D , Mead S , Mosley TH Jr , Bennett DA , Harris TB , Fratiglioni L , Holmes C , de Bruijn RF , Passmore P , Montine TJ , Bettens K , Rotter JI , Brice A , Morgan K , Foroud TM , Kukull WA , Hannequin D , Powell JF , Nalls MA , Ritchie K , Lunetta KL , Kauwe JS , Boerwinkle E , Riemenschneider M , Boada M , Hiltuenen M , Martin ER , Schmidt R , Rujescu D , Wang LS , Dartigues JF , Mayeux R , Tzourio C , Hofman A , Nöthen MM , Graff C , Psaty BM , Jones L , Haines JL , Holmans PA , Lathrop M , Pericak-Vance MA , Launer LJ , Farrer LA , van Duijn CM , Van Broeckhoven C , Moskvina V , Seshadri S , Williams J , Schellenberg GD , Amouyel P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45: , 1452–1458. |
[63] | Hu J , Igarashi A , Kamata M , Nakagawa H ((2001) ) Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta); retards A beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem 276: , 47863–47868. |
[64] | Hemming ML , Selkoe DJ ((2005) ) Amyloid beta -protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem 280: , 37644–37650. |
[65] | Oba R , Igarashi A , Kamata M , Nagata K , Takano S , Nakagawa H ((2005) ) The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide. Eur J Neurosci 21: , 733–740. |
[66] | Toropygin IY , Kugaevskaya EV , Mirgorodskaya OA , Elisseeva YE , Kozmin YP , Popov IA , Nikolaev EN , Makarov AA , Kozin SA ((2008) ) The N-domain of angiotensin-converting enzyme specifically hydrolyzes the Arg-5-His-6 bond of Alzheimer’s Abeta-(1-16) peptide and its isoAsp-7 analogue with different efficiency as evidenced by quantitative matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 22: , 231–239. |
[67] | Zou K , Yamaguchi H , Akatsu H , Sakamoto T , Ko M , Mizoguchi K , Gong JS , Yu W , Yamamoto T , Kosaka K , Yanagisawa K , Michikawa M ((2007) ) Angiotensin-converting enzyme converts amyloid beta-protein 1-42 (Abeta(1-42)) to Abeta(1-40), and its inhibition enhances brain Abeta deposition. J Neurosci 27: , 8628–8635. |
[68] | Zou K , Michikawa M ((2008) ) Angiotensin-converting enzyme as a potential target for treatment of Alzheimer’s disease: Inhibition or activation? Rev Neurosci 19: , 203–212. |
[69] | Eckman EA , Adams SK , Troendle FJ , Stodola BA , Kahn MA , Fauq AH , Xiao HD , Bernstein KE , Eckman CB ((2006) ) Regulation of steady-state beta -amyloid levels in the brain by neprilysin and endothelin-converting enzyme, but not angiotensin-converting enzyme. J Biol Chem 281: , 30471–30478. |
[70] | Hemming ML , Selkoe DJ , Farris W ((2007) ) Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid beta-protein metabolism in mouse models of Alzheimer disease. Neurobiol Dis 26: , 273–281. |
[71] | Yamada K , Uchida S , Takahashi S , Takayama M , Nagata Y , Suzuki N , Shirakura S , Kanda T ((2010) ) Effect of a centrally active angiotensin-converting enzyme inhibitor, perindopril, on cognitive performance in a mouse model of Alzheimer’s disease. Brain Res 1352: , 176–186. |
[72] | Hou DR , Wang Y , Zhou L , Chen K , Tian Y , Song Z , Bao J , Yang QD ((2008) ) Altered angiotensin-converting enzyme and its effects on the brain in a rat model of Alzheimer disease. Chin Med J (Engl) 121: , 2320–2323. |
[73] | Manschot SM , Biessels GJ , Cameron NE , Cotter MA , Kamal A , Kappelle LJ , Gispen WH ((2003) ) Angiotensin converting enzyme inhibition partially prevents deficits in water maze performance, hippocampal synaptic plasticity and cerebral blood flow in streptozotocin-diabetic rats. Brain Res 966: , 274–282. |
[74] | Sink KM , Leng X , Williamson J , Kritchevsky SB , Yaffe K , Kuller L , Yasar S , Atkinson H , Robbins M , Psaty B , Goff DC Jr ((2009) ) Angiotensin-converting enzyme inhibitors and cognitive decline in older adults with hypertension: Results from the cardiovascular health study. Arch Intern Med 169: , 1195–1202. |
[75] | Ferrington L , Miners JS , Palmer LE , Bond SM , Povey JE , Kelly PAT , Love S , Horsburgh KJ , Kehoe PG ((2011) ) Angiotensin II-inhibiting drugs have no effect on intraneuronal Aβ or oligomeric Aβ levels in a triple transgenic mouse model of Alzheimer’s disease. Am J Transl Res 3: , 12. |
[76] | Tota S , Kamat PK , Awasthi H , Singh N , Raghubir R , Nath C , Hanif K ((2009) ) Candesartan improves memory decline in mice: Involvement of AT1 receptors in memory deficit induced by intracerebral streptozotocin. Behav Brain Res 199: , 235–240. |
[77] | Danielyan L , Klein R , Hanson LR , Buadze M , Schwab M , Gleiter CH , Frey WH ((2010) ) Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res 13: , 195–201. |
[78] | Wang J , Ho L , Chen L , Zhao Z , Zhao W , Qian X , Humala N , Seror I , Bartholomew S , Rosendorff C , Pasinetti GM ((2007) ) Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest 117: , 3393–3402. |
[79] | Mogi M , Li JM , Tsukuda K , Iwanami J , Min LJ , Sakata A , Fujita T , Iwai M , Horiuchi M ((2008) ) Telmisartan prevented cognitive decline partly due to PPAR-gamma activation. Biochem Biophys Res Commun 375: , 446–449. |
[80] | Koh EJ , Yoon SJ , Lee SM ((2013) ) Losartan protects liver against ischaemia/reperfusion injury through PPAR-gamma activation and receptor for advanced glycation end-products down-regulation. Br J Pharmacol 169: , 1404–1416. |
[81] | Schupp M , Lee LD , Frost N , Umbreen S , Schmidt B , Unger T , Kintscher U ((2006) ) Regulation of peroxisome proliferator-activated receptor gamma activity by losartan metabolites. Hypertension 47: , 586–589. |
[82] | Horiuchi M , Mogi M ((2011) ) Role of angiotensin II receptor subtype activation in cognitive function and ischaemic brain damage. Br J Pharmacol 163: , 1122–1130. |
[83] | Du J , Sun B , Chen K , Fan L , Wang Z ((2009) ) Antagonist of peroxisome proliferator-activated receptor gamma induces cerebellar amyloid-beta levels and motor dysfunction in APP/PS1 transgenic mice. Biochem Biophys Res Commun 384: , 357–361. |
[84] | Santos MJ , Quintanilla RA , Toro A , Grandy R , Dinamarca MC , Godoy JA , Inestrosa NC ((2005) ) Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J Biol Chem 280: , 41057–41068. |
[85] | Erbe DV , Gartrell K , Zhang YL , Suri V , Kirincich SJ , Will S , Perreault M , Wang S , Tobin JF ((2006) ) Molecular activation of PPARgamma by angiotensin II type 1-receptor antagonists. Vascul Pharmacol 45: , 154–162. |
[86] | Miller BW , Willett KC , Desilets AR ((2011) ) Rosiglitazone and pioglitazone for the treatment of Alzheimer’s disease. Ann Pharmacother 45: , 1416–1424. |
[87] | Inaba S , Iwai M , Furuno M , Tomono Y , Kanno H , Senba I , Okayama H , Mogi M , Higaki J , Horiuchi M ((2009) ) Continuous activation of renin-angiotensin system impairs cognitive function in renin/angiotensinogen transgenic mice. Hypertension 53: , 356–362. |
[88] | Takeda S , Sato N , Takeuchi D , Kurinami H , Shinohara M , Niisato K , Kano M , Ogihara T , Rakugi H , Morishita R ((2009) ) Angiotensin receptor blocker prevented beta-amyloid-induced cognitive impairment associated with recovery of neurovascular coupling. Hypertension 54: , 1345–1352. |
[89] | Harrison C , Acharya KR ((2014) ) ACE for all - a molecular perspective. J Cell Commun Signal 8: , 195–210. |
[90] | Fuchs S , Xiao HD , Hubert C , Michaud A , Campbell DJ , Adams JW , Capecchi MR , Corvol P , Bernstein KE ((2008) ) Angiotensin-converting enzyme C-terminal catalytic domain is the main site of angiotensin I cleavage in vivo. Hypertension 51: , 267–274. |
[91] | Zou K , Maeda T , Watanabe A , Liu J , Liu S , Oba R , Satoh Y , Komano H , Michikawa M ((2009) ) Abeta42-to-Abeta40- and angiotensin-converting activities in different domains of angiotensin-converting enzyme. J Biol Chem 284: , 31914–31920. |
[92] | Sun X , Becker M , Pankow K , Krause E , Ringling M , Beyermann M , Maul B , Walther T , Siems W-E ((2008) ) Catabolic attacks of membrane-bound angiotensin-converting enzyme on the N-terminal part of species-specific amyloid-[beta] peptides. Eur J Pharmacol 588: , 18–25. |
[93] | Zou K , Liu J , Watanabe A , Hiraga S , Liu S , Tanabe C , Maeda T , Terayama Y , Takahashi S , Michikawa M , Komano H ((2013) ) Abeta43 is the earliest-depositing Abeta species in APP transgenic mouse brain and is converted to Abeta41 by two active domains of ACE. Am J Pathol 182: , 2322–2331. |
[94] | Tayler HM , Palmer JC , Thomas TL , Kehoe PG , Paton JF , Love S ((2017) ) Cerebral Abeta40 and systemic hypertension. J Cereb Blood Flow Metab. doi: 10.1177/0271678X1772493 |
[95] | Anthony CS , Corradi HR , Schwager SL , Redelinghuys P , Georgiadis D , Dive V , Acharya KR , Sturrock ED ((2010) ) The N domain of human angiotensin-I-converting enzyme: The role of N-glycosylation and the crystal structure in complex with an N domain-specific phosphinic inhibitor, RXP407. J Biol Chem 285: , 35685–35693. |
[96] | Douglas RG , Sharma RK , Masuyer G , Lubbe L , Zamora I , Acharya KR , Chibale K , Sturrock ED ((2014) ) Fragment-based design for the development of N-domain-selective angiotensin-1-converting enzyme inhibitors. Clin Sci (Lond) 126: , 305–313. |
[97] | Denti P , Sharp SK , Kroger WL , Schwager SL , Mahajan A , Njoroge M , Gibhard L , Smit I , Chibale K , Wiesner L , Sturrock ED , Davies NH ((2014) ) Pharmacokinetic evaluation of lisinopril-tryptophan, a novel C-domain ACE inhibitor. Eur J Pharm Sci 56: , 113–119. |
[98] | Nchinda AT , Chibale K , Redelinghuys P , Sturrock ED ((2006) ) Synthesis and molecular modeling of a lisinopril-tryptophan analogue inhibitor of angiotensin I-converting enzyme. Bioorg Med Chem Lett 16: , 4616–4619. |
[99] | Ehlers MR , Abrie JA , Sturrock ED ((2013) ) C domain-selective inhibition of angiotensin-converting enzyme. J Renin Angiotensin Aldosterone Syst 14: , 189–192. |
[100] | Kalaria RN , Harik SI ((1989) ) Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 53: , 1083–1088. |
[101] | McGeer EG , Singh EA ((1992) ) Angiotensin-converting enzyme in cortical tissue in Alzheimer’s and some other Neurological Diseases. Dementia 3: , 299–303. |
[102] | Zubenko GS , Marquis JK , Volicer L , Direnfeld LK , Langlais PJ , Nixon RA ((1986) ) Cerebrospinal fluid levels of angiotensin-converting enzyme, acetylcholinesterase, and dopamine metabolites in dementia associated with Alzheimer’s disease and Parkinson’s disease: A correlative study. Biol Psychiatry 21: , 1365–1381. |
[103] | Zubenko GS , Volicer L , Direnfeld LK , Freeman M , Langlais PJ , Nixon RA ((1985) ) Cerebrospinal fluid levels of angiotensin-converting enzyme in Alzheimer’s disease, Parkinson’s disease and progressive supranuclear palsy. Brain Res 328: , 215–221. |
[104] | Konings CH , Kuiper MA , Scheltens P , Grijpma AM , van Pelt W , Wolters EC ((1993) ) Re-evaluation of cerebrospinal fluid angiotensin-converting enzyme activity in patients with ‘probable’ Alzheimer’s disease. Eur J Clin Chem Clin Biochem 31: , 495–497. |
[105] | Nielsen HM , Londos E , Minthon L , Janciauskiene SM ((2007) ) Soluble adhesion molecules and angiotensin-converting enzyme in dementia. Neurobiol Dis 26: , 27–35. |
[106] | Savaskan E , Hock C , Olivieri G , Bruttel S , Rosenberg C , Hulette C , Muller-Spahn F ((2001) ) Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer’s dementia. Neurobiol Aging 22: , 541–546. |
[107] | Herrera M , Sparks MA , Alfonso-Pecchio AR , Harrison-Bernard LM , Coffman TM ((2013) ) Lack of specificity of commercial antibodies leads to misidentification of angiotensin type 1 receptor protein. Hypertension 61: , 253–258. |
[108] | Benicky J , Hafko R , Sanchez-Lemus E , Aguilera G , Saavedra JM ((2012) ) Six commercially available angiotensin II AT1 receptor antibodies are non-specific. Cell Mol Neurobiol 32: , 1353–1365. |
[109] | Fyhrquist F , Saijonmaa O ((2008) ) Renin-angiotensin system revisited. J Intern Med 264: , 224–236. |
[110] | Phillips MI , de Oliveira EM ((2008) ) Brain renin angiotensin in disease. J Mol Med 86: , 715–722. |
[111] | Wright JW , Harding JW ((2011) ) Brain renin-angiotensin–a new look at an old system. Prog Neurobiol 95: , 49–67. |
[112] | Haulica I , Bild W , Serban DN ((2005) ) Angiotensin peptides and their pleiotropic actions. J Renin Angiotensin Aldosterone Syst 6: , 121–131. |
[113] | Kanaide H , Ichiki T , Nishimura J , Hirano K ((2003) ) Cellular mechanism of vasoconstriction induced by angiotensin II: It remains to be determined. Circ Res 93: , 1015–1017. |
[114] | Reaux-Le Goazigo A , Iturrioz X , Fassot C , Claperon C , Roques BP , Llorens-Cortes C ((2005) ) Role of angiotensin III in hypertension. Curr Hypertens Rep 7: , 128–134. |
[115] | Reaux A , Iturrioz X , Vazeux G , Fournie-Zaluski MC , David C , Roques BP , Corvol P , Llorens-Cortes C ((2000) ) Aminopeptidase A, which generates one of the main effector peptides of the brain renin-angiotensin system, angiotensin III, has a key role in central control of arterial blood pressure. Biochem Soc Trans 28: , 435–440. |
[116] | Dupont AG , Yang R , Smolders I , Vanderheyden P ((2009) ) IRAP and AT(1) receptor mediated effects of angiotensin IV. J Intern Med 265: , 401–403; author reply 404-405. |
[117] | Lochard N , Thibault G , Silversides DW , Touyz RM , Reudelhuber TL ((2004) ) Chronic production of angiotensin IV in the brain leads to hypertension that is reversible with an angiotensin II AT1 receptor antagonist. Circ Res 94: , 1451–1457. |
[118] | Santos RA , Simoes e Silva AC , Maric C , Silva DM , Machado RP , de Buhr I , Heringer-Walther S , Pinheiro SV , Lopes MT , Bader M , Mendes EP , Lemos VS , Campagnole-Santos MJ , Schultheiss HP , Speth R , Walther T ((2003) ) Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A 100: , 8258–8263. |
[119] | Miners JS , Ashby E , Van Helmond Z , Chalmers KA , Palmer LE , Love S , Kehoe PG ((2008) ) Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer’s disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 34: , 181–193. |
[120] | Kehoe PG , Miners S , Love S ((2009) ) Angiotensins in Alzheimer’s disease - friend or foe? Trends Neurosci 32: , 619–628. |
[121] | Kehoe PG , Wong S , Al Mulhim N , Palmer LE , Miners JS ((2016) ) Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-beta and tau pathology. Alzheimers Res Ther 8: , 50. |
[122] | Mukoyama M , Nakajima M , Horiuchi M , Sasamura H , Pratt RE , Dzau VJ ((1993) ) Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J Biol Chem 268: , 24539–24542. |
[123] | Murphy TJ , Alexander RW , Griendling KK , Runge MS , Bernstein KE ((1991) ) Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature 351: , 233–236. |
[124] | Kehoe PG , Hibbs E , Palmer LE , Miners JS ((2017) ) Angiotensin-III is increased in Alzheimer’s disease in association with amyloid-beta and tau pathology. J Alzheimers Dis 58: , 203–214. |
[125] | Antonios G , Saiepour N , Bouter Y , Richard BC , Paetau A , Verkkoniemi-Ahola A , Lannfelt L , Ingelsson M , Kovacs GG , Pillot T , Wirths O , Bayer TA ((2013) ) N-truncated Abeta starting with position four: Early intraneuronal accumulation and rescue of toxicity using NT4X-167, a novel monoclonal antibody. Acta Neuropathol Commun 1: , 56. |
[126] | Brannstrom K , Ohman A , Nilsson L , Pihl M , Sandblad L , Olofsson A ((2014) ) The N-terminal region of amyloid beta controls the aggregation rate and fibril stability at low pH through a gain of function mechanism. J Am Chem Soc 136: , 10956–10964. |
[127] | Albiston AL , Diwakarla S , Fernando RN , Mountford SJ , Yeatman HR , Morgan B , Pham V , Holien JK , Parker MW , Thompson PE , Chai SY ((2011) ) Identification and development of specific inhibitors for insulin-regulated aminopeptidase as a new class of cognitive enhancers. Br J Pharmacol 164: , 37–47. |
[128] | Albiston AL , Morton CJ , Ng HL , Pham V , Yeatman HR , Ye S , Fernando RN , De Bundel D , Ascher DB , Mendelsohn FA , Parker MW , Chai SY ((2008) ) Identification and characterization of a new cognitive enhancer based on inhibition of insulin-regulated aminopeptidase. FASEB J 22: , 4209–4217. |
[129] | Hajjar I , Brown L , Mack WJ , Chui H ((2012) ) Impact of Angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch Neurol 69: , 1632–1638. |
[130] | Hajjar I , Levey A ((2015) ) Association between angiotensin receptor blockers and longitudinal decline in tau in mild cognitive impairment. JAMA Neurol 72: , 1069–1070. |
[131] | Hellner K , Walther T , Schubert M , Albrecht D ((2005) ) Angiotensin-(1-7) enhances LTP in the hippocampus through the G-protein-coupled receptor Mas. Mol Cell Neurosci 29: , 427–435. |
[132] | Barnes JM , Barnes NM , Costall B , Horovitz ZP , Ironside JW , Naylor RJ , Williams TJ ((1990) ) Angiotensin II inhibits cortical cholinergic function: Implications for cognition. J Cardiovasc Pharmacol 16: , 234–238. |
[133] | Barnes JM , Barnes NM , Costall B , Horovitz ZP , Naylor RJ ((1989) ) Angiotensin II inhibits the release of [3H]acetylcholine from rat entorhinal cortex in vitro. Brain Res 491: , 136–143. |
[134] | Ashby EL , Kehoe PG ((2013) ) Current status of renin-aldosterone angiotensin system-targeting anti-hypertensive drugs as therapeutic options for Alzheimer’s disease. Expert Opin Investig Drugs 22: , 1229–1242. |
[135] | Kivipelto M , Helkala EL , Laakso MP , Hanninen T , Hallikainen M , Alhainen K , Soininen H , Tuomilehto J , Nissinen A ((2001) ) Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 322: , 1447–1451. |
[136] | Launer LJ , Ross GW , Petrovitch H , Masaki K , Foley D , White LR , Havlik RJ ((2000) ) Midlife blood pressure and dementia: The Honolulu-Asia aging study. Neurobiol Aging 21: , 49–55. |
[137] | Skoog I , Lernfelt B , Landahl S , Palmertz B , Andreasson LA , Nilsson L , Persson G , Oden A , Svanborg A ((1996) ) 15-year longitudinal study of blood pressure and dementia. Lancet 347: , 1141–1145. |
[138] | Staessen JA , Richart T , Birkenhager WH ((2007) ) Less atherosclerosis and lower blood pressure for a meaningful life perspective with more brain. Hypertension 49: , 389–400. |
[139] | Akomolafe A , Beiser A , Meigs JB , Au R , Green RC , Farrer LA , Wolf PA , Seshadri S ((2006) ) Diabetes mellitus and risk of developing Alzheimer disease: Results from the Framingham Study. Arch Neurol 63: , 1551–1555. |
[140] | Luchsinger JA ((2010) ) Type 2 diabetes, related conditions, in relation and dementia: An opportunity for prevention? J Alzheimers Dis 20: , 723–736. |
[141] | Hanon O , Haulon S , Lenoir H , Seux ML , Rigaud AS , Safar M , Girerd X , Forette F ((2005) ) Relationship between arterial stiffness and cognitive function in elderly subjects with complaints of memory loss. Stroke 36: , 2193–2197. |
[142] | Ott A , Breteler MM , de Bruyne MC , van Harskamp F , Grobbee DE , Hofman A ((1997) ) Atrial fibrillation and dementia in a population-based study. The Rotterdam Study. Stroke 28: , 316–321. |
[143] | Honig LS , Tang MX , Albert S , Costa R , Luchsinger J , Manly J , Stern Y , Mayeux R ((2003) ) Stroke and the risk of Alzheimer disease. Arch Neurol 60: , 1707–1712. |
[144] | Hanon O , Latour F , Seux ML , Lenoir H , Forette F , Rigaud AS ((2005) ) Evolution of blood pressure in patients with Alzheimer’s disease: A one year survey of a French Cohort (REAL.FR). J Nutr Health Aging 9: , 106–111. |
[145] | Skoog I , Gustafson D ((2006) ) Update on hypertension and Alzheimer’s disease. Neurol Res 28: , 605–611. |
[146] | Kehoe PG , Wilcock GK ((2007) ) Is inhibition of the renin-angiotensin system a new treatment option for Alzheimer’s disease? Lancet Neurol 6: , 373–378. |
[147] | Carey RM ((2015) ) The intrarenal renin-angiotensin system in hypertension. Adv Chronic Kidney Dis 22: , 204–210. |
[148] | Lim HS , MacFadyen RJ , Lip GY ((2004) ) Diabetes mellitus, the renin-angiotensin-aldosterone system, and the heart. Arch Intern Med 164: , 1737–1748. |
[149] | Bangalore S , Fakheri R , Toklu B , Messerli FH ((2016) ) Diabetes mellitus as a compelling indication for use of renin angiotensin system blockers: Systematic review and meta-analysis of randomized trials. BMJ 352: , i438. |
[150] | Sohn SI , Kim CJ ((2014) ) Modulation of renin-angiotensin system and arterial stiffness: Evidence from clinical trials. Curr Hypertens Rev 10: , 37–40. |
[151] | Alhusban A , Fouda AY , Fagan SC ((2016) ) ARBs improve stroke outcome through an AT2-dependent, BDNF-induced proangiogenic and prorecovery response. Neural Regen Res 11: , 912–913. |
[152] | Fouda AY , Artham S , El-Remessy AB , Fagan SC ((2016) ) Renin-angiotensin system as a potential therapeutic target in stroke and retinopathy: Experimental and clinical evidence. Clin Sci (Lond) 130: , 221–238. |
[153] | Wolozin B , Lee A , Lee A , Whitmer R , Kaziz L ((2008) ) Use of angiotensin receptor blockers is associated with lower incidence and progression of Alzheimer’s disease. Alzheimer’s Association International Conference on Alzheimer’s Disease (ICAD) Alzheimer’s & Dementia, McCormick Place, Chicago, p. T118. |
[154] | Davies NM , Kehoe PG , Ben-Shlomo Y , Martin RM ((2011) ) Associations of anti-hypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis 26: , 699–708. |
[155] | Haag MD , Hofman A , Koudstaal PJ , Breteler MM , Stricker BH ((2009) ) Duration of antihypertensive drug use and risk of dementia: A prospective cohort study. Neurology 72: , 1727–1734. |
[156] | Ohrui T , Matsui T , Yamaya M , Arai H , Ebihara S , Maruyama M , Sasaki H ((2004) ) Angiotensin-converting enzyme inhibitors and incidence of Alzheimer’s disease in Japan. J Am Geriatr Soc 52: , 649–650. |
[157] | Ohrui T , Tomita N , Sato-Nakagawa T , Matsui T , Maruyama M , Niwa K , Arai H , Sasaki H ((2004) ) Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology 63: , 1324–1325. |
[158] | Hajjar I , Catoe H , Sixta S , Boland R , Johnson D , Hirth V , Wieland D , Eleazer P ((2005) ) Cross-sectional and longitudinal association between antihypertensive medications and cognitive impairment in an elderly population. J Gerontol A Biol Sci Med Sci 60: , 67–73. |
[159] | Hajjar IM , Keown M , Lewis P , Almor A ((2008) ) Angiotensin converting enzyme inhibitors and cognitive and functional decline in patients with Alzheimer’s disease: An observational study. Am J Alzheimers Dis Other Demen 23: , 77–83. |
[160] | Rozzini L , Chilovi BV , Bertoletti E , Conti M , Del Rio I , Trabucchi M , Padovani A ((2006) ) Angiotensin converting enzyme (ACE) inhibitors modulate the rate of progression of amnestic mild cognitive impairment. Int J Geriatr Psychiatry 21: , 550–555. |
[161] | Rozzini L , Chilovi BV , Trabucchi M , Padovani A ((2008) ) Antihypertensive medications influence the rate of conversion from mild cognitive impairment to Alzheimer disease. Arch Neurol 65: , 993–994. |
[162] | Schneider P , Buerger K , Teipel S , Uspenskaya O , Hartmann O , Hansson O , Minthon L , Rujescu D , Moeller HJ , Zetterberg H , Blennow K , Ernst A , Bergmann A , Hampel H ((2011) ) Antihypertensive therapy is associated with reduced rate of conversion to Alzheimer’s disease in midregional proatrial natriuretic Peptide stratified subjects with mild cognitive impairment. Biol Psychiatry 70: , 145–151. |
[163] | Ellul J , Archer N , Foy CM , Poppe M , Boothby H , Nicholas H , Brown RG , Lovestone S ((2007) ) The effects of commonly prescribed drugs in patients with Alzheimer’s disease on the rate of deterioration. J Neurol Neurosurg Psychiatry 78: , 233–239. |
[164] | Birkenhager WH , Staessen JA ((2006) ) Antihypertensives for prevention of Alzheimer’s disease. Lancet Neurol 5: , 466–468. |
[165] | Feigin V , Ratnasabapathy Y , Anderson C ((2005) ) Does blood pressure lowering treatment prevents dementia or cognitive decline in patients with cardiovascular and cerebrovascular disease? J Neurol Sci 229-230: , 151–155. |
[166] | Louis WJ , Mander AG , Dawson M , O’Callaghan C , Conway EL ((1999) ) Use of computerized neuropsychological tests (CANTAB) to assess cognitive effects of antihypertensive drugs in the elderly. Cambridge Neuropsychological Test Automated Battery. J Hypertens 17: , 1813–1819. |
[167] | Fogari R , Mugellini A , Zoppi A , Derosa G , Pasotti C , Fogari E , Preti P ((2003) ) Influence of losartan and atenolol on memory function in very elderly hypertensive patients. J Hum Hypertens 17: , 781–785. |
[168] | Fogari R , Mugellini A , Zoppi A , Marasi G , Pasotti C , Poletti L , Rinaldi A , Preti P ((2004) ) Effects of valsartan compared with enalapril on blood pressure and cognitive function in elderly patients with essential hypertension. Eur J Clin Pharmacol 59: , 863–868. |
[169] | Forette F , Seux ML , Staessen JA , Thijs L , Birkenhager WH , Babarskiene MR , Babeanu S , Bossini A , Gil-Extremera B , Girerd X , Laks T , Lilov E , Moisseyev V , Tuomilehto J , Vanhanen H , Webster J , Yodfat Y , Fagard R ((1998) ) Prevention of dementia in randomised double-blind placebo-controlled Systolic Hypertension in Europe (Syst-Eur) trial. Lancet 352: , 1347–1351. |
[170] | Forette F , Seux ML , Staessen JA , Thijs L , Babarskiene MR , Babeanu S , Bossini A , Fagard R , Gil-Extremera B , Laks T , Kobalava Z , Sarti C , Tuomilehto J , Vanhanen H , Webster J , Yodfat Y , Birkenhager WH ((2002) ) The prevention of dementia with antihypertensive treatment: New evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Intern Med 162: , 2046–2052. |
[171] | Tzourio C , Anderson C , Chapman N , Woodward M , Neal B , MacMahon S , Chalmers J ((2003) ) Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med 163: , 1069–1075. |
[172] | Skoog I , Lithell H , Hansson L , Elmfeldt D , Hofman A , Olofsson B , Trenkwalder P , Zanchetti A ((2005) ) Effect of baseline cognitive function and antihypertensive treatment on cognitive and cardiovascular outcomes: Study on COgnition and Prognosis in the Elderly (SCOPE). Am J Hypertens 18: , 1052–1059. |
[173] | Yasar S , Xia J , Yao W , Furberg CD , Xue QL , Mercado CI , Fitzpatrick AL , Fried LP , Kawas CH , Sink KM , Williamson JD , DeKosky ST , Carlson MC , Ginkgo Evaluation of Memory (GEM) Study Investigators ((2013) ) Antihypertensive drugs decrease risk of Alzheimer disease: Ginkgo Evaluation of Memory Study. Neurology 81: , 896–903. |
[174] | O’Caoimh R , Healy L , Gao Y , Svendrovski A , Kerins DM , Eustace J , Kehoe PG , Guyatt G , Molloy DW ((2014) ) Effects of centrally acting angiotensin converting enzyme inhibitors on functional decline in patients with Alzheimer’s disease. J Alzheimers Dis 40: , 595–603. |
[175] | O’Caoimh R , Kehoe PG , Molloy DW ((2014) ) Renin Angiotensin aldosterone system inhibition in controlling dementia-related cognitive decline. J Alzheimers Dis 42: (Suppl 4), S575–586. |
[176] | Weiner MF , Bonte FJ , Tintner R , Ford N , Svetlik D , Riall T ((1992) ) ACE inhibitor lacks acute effect on cognition or brain blood flow in Alzheimer’s disease. Drug Dev Res 26: , 467–471. |
[177] | Sudilovsky A , Cutler NR , Sramek JJ , Wardle T , Veroff AE , Mickelson W , Markowitz J , Repetti S ((1993) ) A pilot clinical trial of the angiotensin-converting enzyme inhibitor ceranapril in Alzheimer disease. Alzheimer Dis Assoc Disord 7: , 105–111. |
[178] | Wharton W , Goldstein FC , Zhao L , Steenland K , Levey AI , Hajjar I ((2015) ) Modulation of renin-angiotensin system may slow conversion from mild cognitive impairment to Alzheimer’s disease. J Am Geriatr Soc 63: , 1749–1756. |
[179] | Hajjar I , Hart M , Chen YL , Mack W , Novak V , H CC , Lipsitz L ((2013) ) Antihypertensive therapy and cerebral hemodynamics in executive mild cognitive impairment: Results of a pilot randomized clinical trial. J Am Geriatr Soc 61: , 194–201. |
[180] | Fournier A , Oprisiu-Fournier R , Serot JM , Godefroy O , Achard JM , Faure S , Mazouz H , Temmar M , Albu A , Bordet R , Hanon O , Gueyffier F , Wang J , Black S , Sato N ((2009) ) Prevention of dementia by antihypertensive drugs: How AT1-receptor-blockers and dihydropyridines better prevent dementia in hypertensive patients than thiazides and ACE-inhibitors. Expert Rev Neurother 9: , 1413–1431. |
[181] | Rosenberg PB , Mielke MM , Tschanz J , Cook L , Corcoran C , Hayden KM , Norton M , Rabins PV , Green RC , Welsh-Bohmer KA , Breitner JC , Munger R , Lyketsos CG ((2008) ) Effects of cardiovascular medications on rate of functional decline in Alzheimer disease. Am J Geriatr Psychiatry 16: , 883–892. |
[182] | Lithell H , Hansson L , Skoog I , Elmfeldt D , Hofman A , Olofsson B , Trenkwalder P , Zanchetti A ((2003) ) The Study on Cognition and Prognosis in the Elderly (SCOPE): Principal results of a randomized double-blind intervention trial. J Hypertens 21: , 875–886. |
[183] | Peters R , Beckett N , Forette F , Tuomilehto J , Clarke R , Ritchie C , Waldman A , Walton I , Poulter R , Ma S , Comsa M , Burch L , Fletcher A , Bulpitt C ((2008) ) Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG): A double-blind, placebo controlled trial. Lancet Neurol 7: , 683–689. |
[184] | McGuinness B , Todd S , Passmore P , Bullock R ((2009) ) Blood pressure lowering in patients without prior cerebrovascular disease for prevention of cognitive impairment and dementia. Cochrane Database Syst Rev, CD004034. |
[185] | Staessen JA , Thijs L , Richart T , Odili AN , Birkenhager WH ((2011) ) Placebo-controlled trials of blood pressure-lowering therapies for primary prevention of dementia. Hypertension 57: , e6–7. |
[186] | Iturria-Medina Y , Sotero RC , Toussaint PJ , Mateos-Perez JM , Evans AC , Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7: , 11934. |
[187] | Montagne A , Barnes SR , Sweeney MD , Halliday MR , Sagare AP , Zhao Z , Toga AW , Jacobs RE , Liu CY , Amezcua L , Harrington MG , Chui HC , Law M , Zlokovic BV ((2015) ) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85: , 296–302. |
[188] | Diaz-Ruiz C , Wang J , Ksiezak-Reding H , Ho L , Qian X , Humala N , Thomas S , Martinez-Martin P , Pasinetti GM ((2009) ) Role of hypertension in aggravating aβ neuropathology of AD type and tau-mediated motor impairment. Cardiovasc Psychiatry Neurol 2009: , 107286. |
[189] | Zhu D , Shi J , Zhang Y , Wang B , Liu W , Chen Z , Tong Q ((2011) ) Central angiotensin II stimulation promotes beta amyloid production in Sprague Dawley rats. PLoS One 6: , e16037. |
[190] | Park L , Uekawa K , Garcia-Bonilla L , Koizumi K , Murphy M , Pistik R , Younkin L , Younkin S , Zhou P , Carlson G , Anrather J , Iadecola C ((2017) ) Brain perivascular macrophages initiate the neurovascular dysfunction of Alzheimer Abeta peptides. Circ Res 121: , 258–269. |
[191] | Park L , Zhou P , Pitstick R , Capone C , Anrather J , Norris EH , Younkin L , Younkin S , Carlson G , McEwen BS , Iadecola C ((2008) ) Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A 105: , 1347–1352. |
[192] | Robitaille C , Dai S , Waters C , Loukine L , Bancej C , Quach S , Ellison J , Campbell N , Tu K , Reimer K , Walker R , Smith M , Blais C , Quan H ((2012) ) Diagnosed hypertension in Canada: Incidence, prevalence and associated mortality. CMAJ 184: , E49–56. |
[193] | Mensah GA ((2008) ) Epidemiology of stroke and high blood pressure in Africa. Heart 94: , 697–705. |
[194] | Zlokovic BV ((2005) ) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28: , 202–208. |
[195] | Raghavendra V , Chopra K , Kulkarni SK ((1998) ) Involvement of cholinergic system in losartan-induced facilitation of spatial and short-term working memory. Neuropeptides 32: , 417–421. |
[196] | Kumaran D , Udayabanu M , Kumar M , Aneja R , Katyal A ((2008) ) Involvement of angiotensin converting enzyme in cerebral hypoperfusion induced anterograde memory impairment and cholinergic dysfunction in rats. Neuroscience 155: , 626–639. |
[197] | Decourt B , Lahiri DK , Sabbagh MN ((2017) ) Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr Alzheimer Res 14: , 412–425. |
[198] | Hamdi HK , Castellon R ((2004) ) A genetic variant of ACE increases cell survival: A new paradigm for biology and disease. Biochem Biophys Res Commun 318: , 187–191. |
[199] | Suzuki Y , Ruiz-Ortega M , Lorenzo O , Ruperez M , Esteban V , Egido J ((2003) ) Inflammation and angiotensin II. Int J Biochem Cell Biol 35: , 881–900. |
[200] | Wosik K , Cayrol R , Dodelet-Devillers A , Berthelet F , Bernard M , Moumdjian R , Bouthillier A , Reudelhuber TL , Prat A ((2007) ) Angiotensin II controls occludin function and is required for blood brain barrier maintenance: Relevance to multiple sclerosis. J. Neurosci. 27: , 9032–9042. |
[201] | Berridge MJ ((2011) ) Calcium signalling and Alzheimer’s disease. Neurochem Res 36: , 1149–1156. |
[202] | Gebke E , Muller AR , Jurzak M , Gerstberger R ((1998) ) Angiotensin II-induced calcium signalling in neurons and astrocytes of rat circumventricular organs. Neuroscience 85: , 509–520. |
[203] | Guimaraes PB , Alvarenga EC , Siqueira PD , Paredes-Gamero EJ , Sabatini RA , Morais RL , Reis RI , Santos EL , Teixeira LG , Casarini DE , Martin RP , Shimuta SI , Carmona AK , Nakaie CR , Jasiulionis MG , Ferreira AT , Pesquero JL , Oliveira SM , Bader M , Costa-Neto CM , Pesquero JB ((2011) ) Angiotensin II binding to angiotensin I-converting enzyme triggers calcium signaling. Hypertension 57: , 965–972. |
[204] | Kehoe PG , Passmore PA ((2012) ) The renin-angiotensin system and antihypertensive drugs in Alzheimer’s disease: Current standing of the angiotensin hypothesis? J Alzheimers Dis 30: (Suppl 2), S251–268. |
[205] | Shimizu T , Fukuda H , Murayama S , Izumiyama N , Shirasawa T ((2002) ) Isoaspartate formation at position 23 of amyloid beta peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J Neurosci Res 70: , 451–461. |
[206] | Shin Y , Cho HS , Fukumoto H , Shimizu T , Shirasawa T , Greenberg SM , Rebeck GW ((2003) ) Abeta species, including IsoAsp23 Abeta, in Iowa-type familial cerebral amyloid angiopathy. Acta Neuropathol 105: , 252–258. |
[207] | Tsvetkov PO , Popov IA , Nikolaev EN , Archakov AI , Makarov AA , Kozin SA ((2008) ) Isomerization of the Asp7 residue results in zinc-induced oligomerization of Alzheimer’s disease amyloid beta(1-16) peptide. Chembiochem 9: , 1564–1567. |
[208] | Kuo YM , Webster S , Emmerling MR , De Lima N , Roher AE ((1998) ) Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of A beta peptides of Alzheimer’s disease. Biochim Biophys Acta 1406: , 291–298. |
[209] | Kumar S , Rezaei-Ghaleh N , Terwel D , Thal DR , Richard M , Hoch M , Mc Donald JM , Wullner U , Glebov K , Heneka MT , Walsh DM , Zweckstetter M , Walter J ((2011) ) Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J 30: , 2255–2265. |
[210] | Khachaturian AS , Zandi PP , Lyketsos CG , Hayden KM , Skoog I , Norton MC , Tschanz JT , Mayer LS , Welsh-Bohmer KA , Breitner JC ((2006) ) Antihypertensive medication use and incident Alzheimer disease: The Cache County Study. Arch Neurol 63: , 7. |
[211] | Kehoe PG , Davies NM , Martin RM , Ben-Shlomo Y ((2013) ) Associations of angiotensin targeting antihypertensive drugs with mortality and hospitalization in primary care patients with dementia. J Alzheimers Dis 33: , 999–1008. |
[212] | Furberg CD , Pitt B ((2001) ) Are all angiotensin-converting enzyme inhibitors interchangeable? J Am Coll Cardiol 37: , 1456–1460. |
[213] | Solfrizzi V , Scafato E , Frisardi V , Seripa D , Logroscino G , Kehoe PG , Imbimbo BP , Baldereschi M , Crepaldi G , Di Carlo A , Galluzzo L , Gandin C , Inzitari D , Maggi S , Pilotto A , Panza F ((2013) ) Angiotensin-converting enzyme inhibitors and incidence of mild cognitive impairment. The Italian Longitudinal Study on Aging. Age (Dordr) 35: , 441–453. |
[214] | Hudson M , Humphries K , Tu JV , Behlouli H , Sheppard R , Pilote L ((2007) ) Angiotensin II receptor blockers for the treatment of heart failure: A class effect? Pharmacotherapy 27: , 526–534. |
[215] | Ho JK , Nation DA , lzheimer’s Disease Neuroimaging Initiative ((2017) ) Memory is preserved in older adults taking AT1 receptor blockers. Alzheimers Res Ther 9: , 33. |
[216] | Jochemsen HM , Geerlings MI , Grool AM , Vincken KL , Mali WP , van der Graaf Y , Muller M ((2012) ) Angiotensin-converting enzyme and progression of white matter lesions and brain atrophy–the SMART-MR study. J Alzheimers Dis 29: , 39–49. |
[217] | Jochemsen HM , Teunissen CE , Ashby EL , van der Flier WM , Jones RE , Geerlings MI , Scheltens P , Kehoe PG , Muller M ((2014) ) The association of angiotensin-converting enzyme with biomarkers for Alzheimer’s disease. Alzheimers Res Ther 6: , 27. |
[218] | Li NC , Lee A , Whitmer RA , Kivipelto M , Lawler E , Kazis LE , Wolozin B ((2010) ) Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: Prospective cohort analysis. BMJ 340: , b5465. |
[219] | Cushman DW , Wang FL , Fung WC , Grover GJ , Harvey CM , Scalese RJ , Mitch SL , DeForrest JM ((1989) ) Comparisons vitro, ex vivo, and in vivo of the actions of seven structurally diverse inhibitors of angiotensin converting enzyme (ACE). Br J Clin Pharmacol 28: (Suppl 2) 115S–130S; discussion 130S-131S. |
[220] | Cushman DW , Wang FL , Fung WC , Harvey CM , DeForrest JM ((1989) ) Differentiation of angiotensin-converting enzyme (ACE) inhibitors by their selective inhibition of ACE in physiologically important target organs. Am J Hypertens 2: , 294–306. |
[221] | Gohlke P , Scholkens B , Henning R , Urbach H , Unger T ((1989) ) Inhibition of converting enzyme in brain tissue and cerebrospinal fluid of rats following chronic oral treatment with the converting enzyme inhibitors ramipril and Hoe 288. J Cardiovasc Pharmacol 14: (Suppl 4), S32–36. |
[222] | Jackson B , Cubela R , Sakaguchi K , Johnston CI ((1987) ) Pharmacokinetics of angiotensin converting enzyme inhibition in tissues following oral lisinopril: Studies in the rat using quantitative radioinhibitor binding. Clin Exp Pharmacol Physiol 14: , 343–347. |
[223] | Tan J , Wang JM , Leenen FH ((2005) ) Inhibition of brain angiotensin-converting enzyme by peripheral administration of trandolapril versus lisinopril in Wistar rats. Am J Hypertens 18: , 158–164. |
[224] | Fabris B , Chen BZ , Pupic V , Perich R , Johnston CI ((1990) ) Inhibition of angiotensin-converting enzyme (ACE) in plasma and tissue. J Cardiovasc Pharmacol 15: (Suppl 2), S6–13. |
[225] | Hu K , Bahner U , Gaudron P , Palkovits M , Ring M , Fehle A , Kruse B , Ertl G ((2001) ) Chronic effects of ACE-inhibition (quinapril) and angiotensin-II-type-1 receptor blockade (losartan) on atrial natriuretic peptide in brain nuclei of rats with experimental myocardial infarction. Basic Res Cardiol 96: , 258–266. |
[226] | Johnston CI , Fabris B , Yamada H , Mendelsohn FA , Cubela R , Sivell D , Jackson B ((1989) ) Comparative studies of tissue inhibition by angiotensin converting enzyme inhibitors. J Hypertens Suppl 7: , S11–16. |
[227] | Jouquey S , Mathieu MN , Hamon G , Chevillard C ((1995) ) Effect of chronic treatment with trandolapril or enalapril on brain ACE activity in spontaneously hypertensive rats. Neuropharmacology 34: , 1689–1692. |
[228] | Sadoshima S , Fujii K , Ooboshi H , Ibayashi S , Fujishima M ((1993) ) Angiotensin converting enzyme inhibitors attenuate ischemic brain metabolism in hypertensive rats. Stroke 24: , 1561–1566; discussion 1566-1567. |
[229] | Sakaguchi K , Chai SY , Jackson B , Johnston CI , Mendelsohn FA ((1988) ) Differential angiotensin-converting enzyme inhibition in brain after oral administration of perindopril demonstrated by quantitative in vitro autoradiography. Neuroendocrinology 48: , 223–228. |
[230] | Sakaguchi K , Chai SY , Jackson B , Johnston CI , Mendelsohn FA ((1988) ) Inhibition of tissue angiotensin converting enzyme. Quantitation by autoradiography. Hypertension 11: , 230–238. |
[231] | Golding BJ , Overall AD , Brown G , Gard PR ((2010) ) Strain differences in the effects of angiotensin IV on mouse cognition. Eur J Pharmacol 641: , 154–159. |
[232] | Chai SY , Yeatman HR , Parker MW , Ascher DB , Thompson PE , Mulvey HT , Albiston AL ((2008) ) Development of cognitive enhancers based on inhibition of insulin-regulated aminopeptidase. BMC Neurosci 9: (Suppl 2), S14. |
[233] | Gard PR ((2008) ) Cognitive-enhancing effects of angiotensin IV. BMC Neurosci 9: (Suppl 2), S15. |
[234] | Gallo-Payet N , Guimond MO , Bilodeau L , Wallinder C , Alterman M , Hallberg A ((2011) ) Angiotensin II, a neuropeptide at the frontier between endocrinology and neuroscience: Is there a link between the angiotensin II type 2 receptor and Alzheimer’s disease? Front Endocrinol (Lausanne) 2: , 17. |
[235] | Guimond MO , Gallo-Payet N ((2012) ) How does angiotensin AT(2) receptor activation help neuronal differentiation and improve neuronal pathological situations? Front Endocrinol (Lausanne) 3: , 164. |
[236] | Wharton W , Goldstein FC , Tansey MG , Brown AL , Tharwani SD , Verble DD , Cintron A , Kehoe PG ((2018) ) Rationale and design of the mechanistic potential of antihypertensives in preclinical Alzheimer’s (HEART) trial. J Alzheimers Dis 61: , 815–824. |

