Article Text

Abstract
Background—The development of clinical disease after infection with Helicobacter pylori has been reported to be associated with expression of the cagA gene. Recently, it has been shown that cagA is part of a multigene locus, described as the cag pathogenicity island (PAI). The role of this region in determining clinical outcome remains to be established.
Aims—To investigate whether the presence ofcagA is always associated with the presence of the complete cag PAI and to evaluate the distribution of selected cag genes in 73 H pylori strains isolated from patients in France.
Methods—Clinical strains of H pyloriwere screened for selected genes of the cag PAI by polymerase chain reaction and colony hybridisation.
Results—Of 64 strains that harboured thecagA gene, 57 (89%) also contained the entirecag PAI. The entire cag PAI was found in 85% (48/56) and 53% (9/17) of duodenal ulcer and non-ulcer dyspepsia isolates, respectively. Eight strains had deletions within thecag PAI, including deletion of the cagA gene in one isolate; the deletions were not associated with the insertion sequence IS605. Of eight strains lacking the cag PAI, four were isolated from patients with duodenal ulcer.
Conclusion—The cag PAI is not a uniform, conserved entity. Although the presence of thecag PAI is highly associated with duodenal ulcer, the clinical outcome of infection with H pylori is not reliably predicted by any gene of the cag PAI.
- duodenal ulcer
- Helicobacter pylori
- cag pathogenicity island
- non-ulcer dyspepsia.
Statistics from Altmetric.com
Helicobacter pylori is a Gram negative, microaerophilic, spiral bacterium that colonises the human stomach.1 Infection with H pylori is associated with chronic superficial gastritis and peptic ulceration,2 and epidemiological evidence of a link with gastric adenocarcinoma and mucosa associated lymphoid tissue (MALT) lymphoma3 ,4 has resulted in classification of the organism as a group I carcinogen.5
CagA was first described as an immunodominant antigen with a molecular mass of 120 kDa.6 This antigen is expressed by the majority of vacuolating cytotoxin producing isolates and consequently the gene encoding this high molecular weight antigen was designated thecagA gene (cytotoxin associated gene).7 ,8The function of CagA remains unclear, as although it is frequently associated with cytotoxin production and the induction of interleukin 8 (IL-8) by gastric epithelial cells, neither of these features are affected by inactivation of cagA.9-11Despite this, a number of studies have suggested that CagA is a useful marker for the more virulent strains that are associated with severe gastroduodenal disease.12 ,13 H pyloristrains that express CagA cause more extensive inflammation of the gastric mucosa14-17 and infections with CagA positive strains have been reported to be more likely to result in peptic ulceration,7 ,18 atrophic gastritis19 and gastric adenocarcinoma.20 ,21 However, studies recently performed in some parts of the world have cast doubt on this association, reporting minimal correlation between the expression of CagA and either inflammation or clinical disease.22-25
Recently, the multigenic locus upstream of cagA was characterised in the strain CCUG 17874 (also designated NCTC 11638) and was found to have the typical features of a pathogenicity island.26 In this strain, the locus is divided into two subregions, cagI and cagII, separated by intervening chromosomal DNA and a sequence reminiscent of an insertion sequence, designated IS605 (fig 1). The latter encodes two putative transposases, TnpA and TnpB, and full length or partial copies of this insertion sequence may also be present elsewhere in the chromosome.26 ,27

Schematic representation of the cag pathogenicity island of H pylori as previously published for the strains NCTC 11638, CCUG 17874, and 26695.26 ,27 Strain 26695 contains a single, contiguous cag PAI, whereas in strain CCUG 17874 (also designated NCTC 11638) the PAI is divided into two regions, cagI and cagII, by intervening chromosomal DNA and the insertion sequence IS605.26 ,27 Arrows represent predicted open reading frames; shaded arrows represent genes targeted in this study.
Based on the analysis of a series of isolates, which included strains with deletions within the cag PAI, Censini et al proposed that subpopulations with intermediate virulence arose after integration of IS605 into the chromosome and subsequent rearrangements and deletions within the cagPAI.26 In the recently sequenced strain 26695 thecag PAI was found as a contiguous unit.27 Thecag PAI encodes proteins with similarity to components of bacterial secretory pathways, including the type IV system, and it has been proposed that the region encodes a secretion system for the export of virulence determinants.26 ,28 Mutation of several of the predicted coding regions of the cagI region resulted in abolition of IL-8 induction, increased haemolytic activity and altered duplication times in liquid culture.26 ,29 The induction of pedestal structures and host protein tyrosine phosphorylation, observed in in vitro assays when H pyloricontacts epithelial cells,30 is also abolished by mutations mapping to the cag region, which suggests it may also export macromolecules involved in the H pylori–host cell interaction.31
To date, only the expression of the CagA antigen, as measured by the detection of antibodies to CagA in H pylori infected patients, or the presence of the cagA gene, as determined by polymerase chain reaction (PCR) or hybridisation, have been investigated in large scale studies of clinical isolates. We therefore decided to investigate a large number of H pylori strains (i) to examine whether the presence of cagA was always associated with the presence of a complete cag PAI and (ii) to study the distribution of several cag genes in relation to the clinical presentation of the patients from whom the strains originated.
Methods
MATERIALS
Antral biopsy specimens were taken from patients consulting for duodenal ulcer disease (n=62) and non-ulcer dyspepsia (NUD) (n=20) in 30 different centres in France. None of the patients was receiving antisecretory or non-steroidal anti-inflammatory drugs. Biopsy samples were ground into brucella broth to disperse the bacteria and the ground material was inoculated onto Wilkins Chalgren plates (Oxoid, Lyon, France) supplemented with 10% human blood and the following antibiotics: vancomycin (10 mg/l), cefsulodine (5 mg/l), and cyclohexamide (100 mg/l). Plates were incubated at 37°C under microaerobic conditions for seven days. For each biopsy specimen all the colonies that grew on selective media were pooled and resuspended into 100 to 500 μl of distilled water, to give a suspension with an A600 of 0.5. The suspension was boiled for five minutes, cooled on ice, and centrifuged for five minutes at 12 000 ×g. Supernatants were collected and frozen at −20°C until processed for gene amplification experiments. Pellets were independently stored at −20°C for colony hybridisation. Lysates from the strains CCUG 17874, 26695, and 85P were prepared in a similar fashion for use as controls.26 ,27 ,32
POLYMERASE CHAIN REACTION
Table 1 shows the nucleotide sequences of the primers used for the different amplification reactions; they were derived from the published sequences of the H pylori cagregion (GenBank accession numbers U601176, AC000108 and AE000511) and the phosphoglucosamine mutase gene, glmM (previously designated ureC).33 PCR was performed as follows: target DNA (the bacterial lysate) was heat denatured prior to the addition to 50 μl of amplification reaction containing 50 pmol of each primer, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.01% (w/v) gelatin, 0.2 mM of each deoxynucleotide (Pharmacia Biotech, St-Quentin-Yvelines, France) and 2.5 units of Taq polymerase (Amersham, Little Chalfont, UK). Gene amplification was carried out through 30 consecutive cycles consisting of a denaturation step of 94°C for two minutes, a primer annealing step ranging from 50–56°C for two minutes (depending on the melting temperature of the various primers) and an extension step at 72°C for two minutes. At the end of the reaction 40 μl of each sample was loaded onto a 1.5% agarose gel containing 0.4 μg/ml ethidium bromide in acetate buffer.
Oligonucleotide primers used in this study
COLONY HYBRIDISATION
Bacterial pellets stored at −20°C were thawed and resuspended in 500 μl distilled water. A volume of 200 μl was filtered through a nylon membrane (Amersham) using a dot blot 96 well apparatus filtration system. To lyse the bacteria, each membrane was placed on 3MM chromatography paper (Whatman International Ltd, Maidstone, UK) saturated with NaOH (0.5 M) for 10 minutes at room temperature. The membranes were then transferred successively onto 3MM paper saturated with neutralisation buffer (1 M ammonium acetate; 0.02 M NaOH) twice for two minutes and finally once for 10 minutes. The membranes were air dried and then baked for two hours at 80°C. The nine probes were generated from chromosomal DNA of H pylori 85P by PCR using the pairs of oligonucleotides listed in table 1 (CAG21 and CAG22 were used for open reading frame 6). The amplified DNA fragments were purified by migration on an agarose gel. Bands of the appropriate size were eluted from the gel and the DNA was extracted and purified by passage through an Elutip minicolumn (Schleicher and Schuell, Dassel, Germany). The DNA was labelled by random priming using the Megaprime DNA labelling system (Amersham) and α-32dCTP (Amersham). The membranes were prehybridised in 50% formamide solution (20 ml per membrane) at 42°C for six hours. The prehybridisation mixture was replaced by the same fresh solution to which 20 μl of the probe was added. After 12 hours of incubation at 42°C under rotative agitation, the membranes were washed three times in 2 × SSC (0.3 M NaCl, 0.03 M sodium citrate)/0.1% sodium dodecyl sulphate at 68°C, were wrapped in plastic films and autoradiography was performed with xray film (Hyperfilm, Amersham) for 18 to 48 hours. The membranes were reused after stripping of previously hybridised probes with 0.1 M NaOH.
Results
DEFINING CagA STATUS
To account for the possibility that patients might have been infected with multiple strains of H pylori all colonies that grew from each biopsy sample were collected and pooled. All the lysates were coded and the analysis was performed blindly. Initially, the 82 lysates were tested in parallel with two sets of oligonucleotides, HP1 and HP2, and CAG1 and CAG2. The HP1 and HP2 oligonucleotides were used to calibrate the lysates and to confirm the presence of H pylori DNA. Figure 2A shows that when theglmM gene was targeted, a 294 bp PCR product was visualised as a unique and homogenous band, which had a similar intensity in all lanes. All lysates which consistently gave a negative amplification with HP1 and HP2 were considered of insufficient quality to allow detection of the selected genes by PCR and were removed from the study (n=9). The CAG1 and CAG2 oligonucleotides were designed to detect the presence of the cagA gene and amplified a 404 bp product. The cagA status of the remaining 73 lysates was determined by PCR (fig 2A) and colony hybridisation (fig 2B) and lysates were designated cagA negative if they were negative for both these investigations. All others were designatedcagA positive lysates. The correlation between results obtained for cagA by PCR and colony hybridisation was 98.7%.

{kind=link}
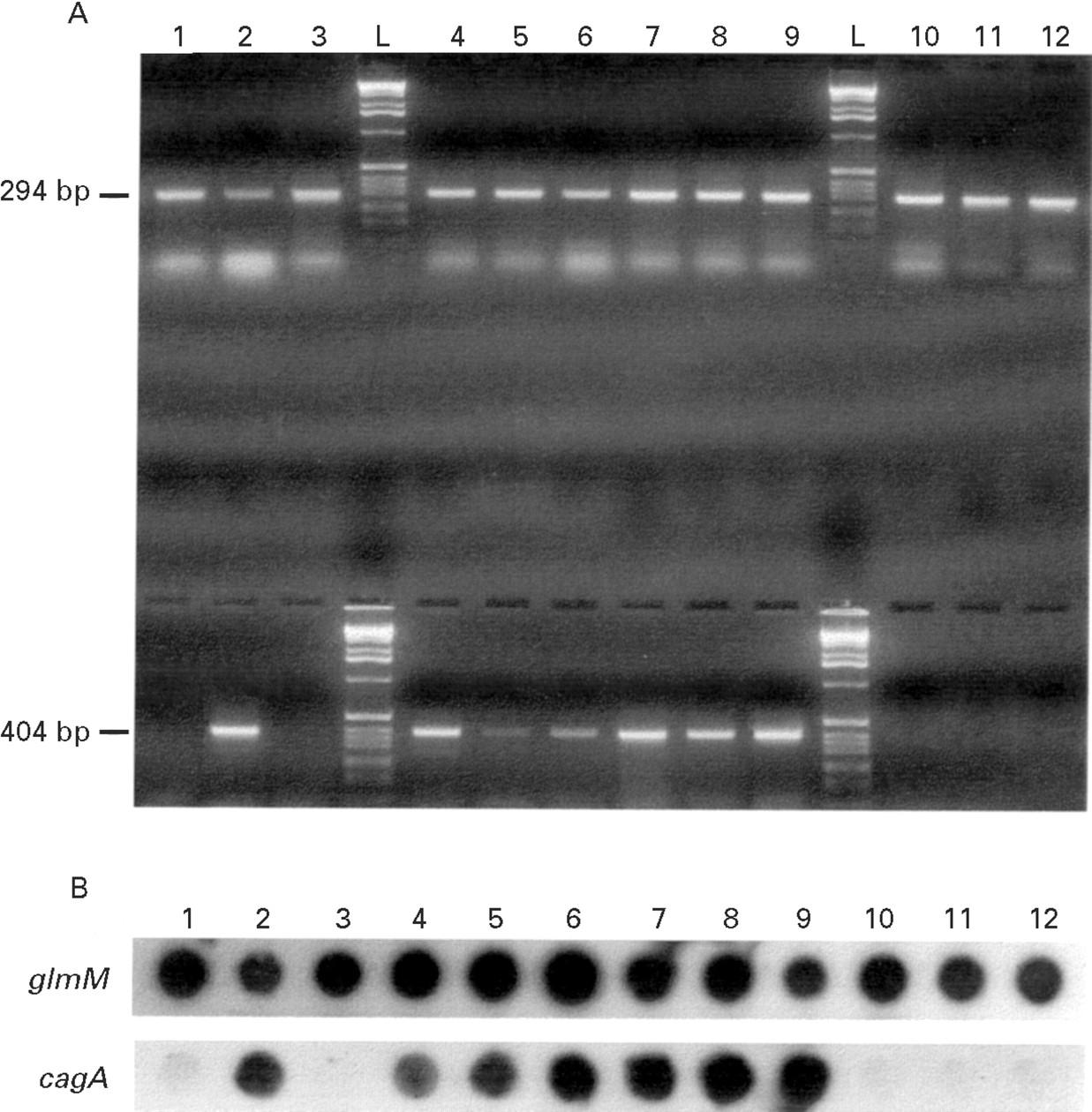{kind=link}
(A) Example of detection of PCR products by agarose gel electrophoresis and ethidium bromide staining. Strains were initially tested for the presence of glmM (upper lanes) and cagA (lower lanes). Lanes 1 to 12 represent strains 26 to 37; lanes L contain a molecular weight marker (Gibco/BRL Ltd, Paisley, UK). (B) Representative colony hybridisation to detect the presence of glmM and cagA. Dots 1 to 12 represent strains 26 to 37. The photocomposition of the figure was obtained from the original Polaroid film plus the autoradiographs from the colony hybridisations with a Studioscan IIsi scanner (AGFA, Mortsel, Belgium). After the initial image was scanned and saved as a PICT file, the file was opened in Adobe Photoshop, version 3.0 (Adobe system Inc. Mountain View, California, USA).
DISTRIBUTION OF SELECTED GENES WITHIN THEcag PAI
Two criteria were used to select the genes of the cagPAI targeted in this study: (i) representative spacing along the 40 kilobase cag PAI and (ii) either the ability to induce IL-8 secretion by gastric epithelial cells or similarity to recognised virulence factors in other bacteria (fig 1). Three loci were selected in the cagI region; cagA,cagE (induces IL-8 and similarity to thevirB4 gene of Agrobacterium tumefaciens 34) and cagM (induces IL-8 and similarity to the hook associated protein type 3 of Vibrio parahaemolyticus 35). Four loci were chosen from thecagII region; cagT (similarity to IPAC surface antigen of Shigella flexneri 36); open reading frame (ORF) 13 (similarity tovirB10 34), ORF10 (similarity tovirD4 37) and ORF6 (the start of thecag PAI, GenBank accession number AC000108). In addition, both tnpA and tnpB genes of the insertion sequence IS605 were selected.
The presence of these selected genes within the cag PAI of the 73 strains was initially determined by PCR amplification. Eight of the oligonucleotide primer pairs selected to target these loci were found to have 100% homology to the equivalent sequences in the recently sequenced H pylori strain 26695.27The oligonucleotide CAGl9, which targeted ORF6, was not found in strain 26695. This was due to a 97 bp deletion at the 5′ end of this gene in strain 26695 compared with NCTC 11638.27 The oligonucleotide primers CAG21 and CAG22 were therefore designed to amplify a region of ORF6 known to be present in all currently sequenced strains. These were used to test the three clinical strains which were negative for ORF6 with CAG19 and CAG20, but which were positive for all the other genes in the cag PAI. These three strains all contained this truncated version of ORF6 (data not shown). Although we used conserved oligonucleotide primers, interstrain variation in the sequences targeted by these primers could still have resulted in non-detection of some of the selected genes in certain strains. The presence of the selected genes was therefore also determined by colony hybridisation. The correlation between the results obtained by PCR and colony hybridisation was 97.9% for the genes of the cagPAI, 98.6% for tnpA and 82.2% for tnpB.
Table 2 shows the detection of the seven selected genes of thecag PAI in strains of H pylori. All of the selected genes of the cag PAI were detected in 57 (78%) of the strains tested. In 47 of these, the IS605 insertion sequence was not found and it can therefore be assumed that the cag PAI was present as a single and entire block of genes in at least 64% of the analysed isolates. Of the 57 strains that contained the entire genetic information of the cag PAI, 10 (18%) contained either partial or complete copies of IS605. We wished to determine whether the elements of IS605 were localised within thecag PAI or elsewhere in the chromosome, and also whether the overall structure of the cag PAI of these isolates was similar to that of the first cag PAI described by Censiniet al for strain CCUG 1787426 (in which thecag PAI is divided into two regions, cagI andcagII, by insertion of IS605 between cagQ andcagS as shown in fig 1). We therefore determined the distance between the CAG7 and CAG12 oligonucleotide primers (internal to cagS and cagQ respectively) by PCR gene amplification. In all 10 isolates a 532 bp PCR product was detected, indicating that cagQ and cagS were adjacent to each other and not disrupted by an IS605 element (data not shown). In these strains the IS605 element is most likely to be localised outside the cag PAI, although we cannot exclude its localisation within one of the open reading frames not targeted by this study.
Detection of selected genes of the cag PAI in clinical strains of H pylori
Of the 73 isolates examined, eight contained only part of thecag PAI. Four lacked the genetic information spanning ORF6 to ORF19 (cagT), but had the information extending fromcagM to cagA. Three of the four had no IS605 element within their genome and hence these rearrangements must have arisen by a mechanism independent of this insertion sequence. For strain 47, the other of the five isolates, no amplification product was detected using either the CAG7 and CAG12 or CAG7 and CAG10 oligonucleotide primers (data not shown), indicating that the deletion extended to the cagQ gene and was not the result of rearrangements induced by the presence of an IS605 element between thecagS and cagQ genes. Three isolates, whether or not containing the IS605 element within their genome, had huge deletions within the cag PAI. These only retained all or part of the information between cagN and cagAand had therefore lost most of the genes of the cag PAI. In contrast, one isolate retained the genes extending at least from ORF6 to cagQ, but had lost those between cagMand cagA; again no elements of IS605 were found in this strain. Finally, in eight of the 73 isolates we could not detect any of the genes of the cag PAI nor of the IS605 insertion sequence.
When analysing the data by clinical presentation (table 3), there was a clear association between ulcerogenic strains and the presence of the entire cag PAI, with 48 of 56 (85%) duodenal ulcer strains containing the entire genetic information encoded by thecag PAI, compared with nine (53%) of 17 NUD strains. A number of patients with peptic ulcer disease harboured strains with partial deletions of the cag PAI. In addition, there were four duoenal ulcer strains in which the entire cag PAI was absent. In 11% of the isolates that harboured the cagAgene, the genetic information encoded by the cag PAI was incomplete, thus rendering the PAI non-functional as a conserved unit.
Distribution of selected genes of the cag PAI and clinical presentation
Discussion
The recently described locus upstream of cagA is the first region with the features of a pathogenicity island to be described in H pylori and to date it has been assumed thatcagA is a marker for this group of genes. In this study the presence of cagA was associated with the presence of the whole cag PAI in only 89% of strains. Like Cesiniet al,26 we found a number of strains which had deletions within the PAI, including one isolate in whichcagA was absent. None of the genes we tested, includingcagA, proved to be reliable markers for the presence of the entire island and it appears that the cag PAI is not a uniform, conserved entity. The presence of cagA, as detected by PCR or hybridisation, or the expression of CagA, cannot therefore be considered an absolute marker for the presence of thecag PAI as a complete set of genes associated with pathogenicity.
Clinical isolates of H pylori have previously been classified into two broad families on the basis of the presence ofcagA and the expression of the vacuolating cytotoxin (VacA).13 It has been proposed that strains associated with more severe gastrointestinal diseases (type I) express both CagA and VacA and exhibit vacuolating activity, while those with attenuated virulence (type II) lack the cagA gene and have avacA that is silent or encodes for a non-toxic but immunoreactive molecule.7 ,13 ,18 It has also been suggested recently that the cag PAI encodes factors important for virulence and Censini et al reported that this region was unique to strains associated with more severe gastrointestinal disease.26 They also proposed that integration of IS605 was associated with subsequent rearrangements and deletions within the cag PAI.26 ,38 We found that most of the strains we tested contained cagI andcagII regions that were not disrupted by an IS605 element and were more likely fused together, and this form of thecag PAI was present in 48 (85%) of 56 duodenal ulcer strains. Importantly, although our series contained relatively few NUD strains, 53% of these also contained the entire cag PAI, suggesting that this region is not restricted to strains associated with severe gastroduodenal disease at the time of presentation. In addition, the strain that contained only the genes of thecagII region, two strains with an apparent deletion upstream of cagE, and four strains negative for the entire PAI were isolated from patients with duodenal ulcers. It is recognised that the presence of cagA is not restricted to strains isolated from patients with duodenal ulcers.14 ,18 Our results provide evidence that the same is true for the entirecag PAI and that this region is not an essential requirement for duodenal ulcer formation. Although it is recognised that heterogeneity of clinical presentation in H pyloriinfected patients may be due to a mixed infection with CagA positive and CagA negative strains,39 we were careful to include in our analysis all colonies that grew from each biopsy specimen in an attempt to account for the possibility of mixed infection. More recently it has been argued that instability and loss of thecag region after infection is established is a better explanation for this phenomenon than infection with multiple strains.38 Our results suggest that either the PAI is not exclusively found in ulcerogenic strains, or the region is highly unstable and easily lost once infection is established.
In contrast to previous studies, we found the entire insertion sequence IS605 in only 11 strains, whereas a further two strains contained one of either tnpA or tnpB. In addition the presence of IS605 was not associated with deletions within thecag PAI. Three of the strains which only contained the genes of the cagI region, the strain that only contained the genes of the cagII region and one of the strains that only contained cagA and cagE did not harbour either of these transposases. The deletions in these strains cannot have arisen as a result of IS605 and must have been mediated by some other mechanism. This implies that the role of this insertion sequence in the evolution of the cag PAI requires further evaluation.
In summary, we have demonstrated that the cag PAI is highly associated with duodenal ulceration, but is not restricted to strains causing severe gastroduodenal disease. We have also shown that patients with peptic ulcer disease may harbour strains with partial or complete deletions of the cag PAI. This suggests that although the cag PAI may be involved in establishing clinical disease, other factors important for pathogenicity remain to be identified. Finally, none of the 73 isolates analysed contained thecag PAI structure (cagI andcagII interrupted by IS605) reported to be present in the CCUG 17874 isolate. This may reflect differences in the structure of the cag PAI in strains isolated from different geographical locations.
Acknowledgments
PJ Jenks was supported by a Research Training Fellowship in Medical Microbiology from The Wellcome Trust, UK. We thank Cecile Birac from the Pellegrin Hospital, Bordeaux, for processing the clinical strains.