Article Text

Abstract
Aims—To assess the validity and practicality of real time polymerase chain reaction (PCR) for human papillomavirus (HPV) testing in combination with liquid based cytology samples for cervical screening.
Methods—Real time PCR using consensus (GP5+/6+) and type specific primers was developed to detect genital HPV types. This provides rapid, efficient amplification followed by denaturation of the product and computer analysis of the kinetics data that are generated. Liquid based cytology samples were obtained from patients attending routine cervical screening clinics. DNA was extracted from the residual cellular suspension after cytology using spin columns.
Results—Real time PCR successfully distinguished between HPV-16 and HPV-18 on the basis of amplification with consensus primers followed by DNA melting temperature (Tm) analysis. Sensitivities of one to 10 copies of HPV-16 (mean Tm = 79.4°C; 2 SD, 0.8) and four to 40 copies of HPV-18 (mean Tm = 80.4°C; 2 SD, 0.4) were obtained. In a mixed population of SiHa and HeLa cells containing known copy numbers of HPV-16 and HPV-18 genomes, HPV-16 and HPV-18 products were clearly separated by Tm analysis in mixtures varying from equivalence to 1/1000. Together with detailed melt analysis, type specific primers from the same region of the L1 gene confirmed the differential ability of this system. The method was applied to 100 liquid based cytology samples where HPV status using conventional GP5+/6+ PCR was already known. There was 95% agreement between the methods, with 55 positives detected by conventional PCR and 59 with real time PCR. The method was then tested on 200 routine liquid based cytology samples. Approximately 10% were positive by real time PCR, most of which were classified as HPV-16 by detailed melt analysis. Thirteen (6.8%) HPV positives were identified in 189 samples showing no evidence of cervical cytological abnormality.
Conclusions—Real time PCR is a rapid, efficient method for the detection of HPV with the separation of HPV-16 and HPV-18 on the basis of differential Tm. Preliminary results suggest it could prove useful if HPV testing is added to cervical screening programmes.
- real time polymerase chain reaction
- cervical screening
- human papillomavirus types 16 and 18
Statistics from Altmetric.com
More than 40 types of human papillomavirus (HPV) infect the genital epithelium and several high risk types including HPV types 16, 18, 31, 33, and 45 are found in almost all cases of high grade cervical intraepithelial neoplasia and cervical cancer.1, 2 In Europe, the most prevalent type is HPV-16,1 but there are several reports that HPV-18 infection can lead to the development of more clinically aggressive disease.3–5
Laboratory diagnosis of HPV infection is dependent upon molecular techniques such as DNA hybridisation or nucleic acid amplification. Several polymerase chain reaction (PCR) methods have been developed to detect a broad spectrum of mucosotropic HPV types using either degenerate or consensus primers.6–9 A second generation commercial hybridisation assay, Hybrid Capture™ (HCA II), is also available for the detection of HPV DNA in cervical swab samples,10 and has been used widely in epidemiological studies.11–13 However, both consensus PCR and HCA II have important limitations. They are costly and labour intensive and, without additional procedures, neither technique can differentiate between individual types or detect infection with more than one type. Furthermore, HPV infections are often transient, frequently cleared by immunocompetent people, and require interaction with cofactors for the progression of disease. Thus, the development of highly sensitive detection tests for high risk HPV raises problems of clinical interpretation.
The potential use of HPV testing in cervical screening programmes is dependent on a rapid sensitive test that can distinguish high risk HPV types present in clinical samples. In most conventional PCR assays, amplification is performed by automated temperature cycling, but product analysis requires a subsequent manual operation.
Rapid real time PCR can distinguish closely related sequences on the basis of amplification followed by DNA melting temperature analysis. The commercial LightCycler (Idaho Technology Inc, supplied by BioGene Ltd, Kimbolton, Cambridgeshire, UK) combines simultaneous PCR amplification with sophisticated computer analysis of the kinetics data generated. The use of air as a circulating medium during PCR cycling allows rapid temperature control and thus a significant reduction in testing time (typically 40 cycles in 25 minutes). The use of fine capillaries of borosilicate glass provides efficient heat transfer and by acting as wave guides facilitates sensitive fluorimetry and enhances the efficiency of the amplification. The amplification mix contains a fluorescent dye, SYBR Green I™, which binds to the minor groove of double stranded DNA and emits light on excitation.14 Thus, as the PCR product accumulates, fluorescence increases. On denaturation of the product, SYBR Green I is released and fluorescence rapidly decreases. Because the melting curve of DNA is dependent on sequence, length, and GC content, PCR products can be distinguished by their melting curves. The determination of melting curves can be carried out on each sample after amplification without opening the reaction vessels.
We report the differentiation of HPV-16 and HPV-18 in mixed cell populations using GP5+/6+ consensus primers,9 with confirmation using type specific modifications of these primers, and the application of this method to clinical specimens.
Materials and methods
POSITIVE CONTROL MATERIAL
HPV containing cell lines
The cell lines, SiHa and HeLa, which contain one to two copies of HPV-16 DNA and 30–40 copies of HPV-18 DNA/cell, respectively,15 were grown as monolayers and passaged at regular intervals using standard cell culture techniques. Cells were removed from the plastic with gentle trypsinisation, counted, and the DNA extracted from a known number of cells in 200 μl of phosphate buffered saline (PBS) using spin columns (Qiagen DNA mini kit; Qiagen Ltd, Lewes, West Sussex, UK) in accordance with the manufacturer's tissue protocol.
Cloned material
Cloned DNAs of HPV types 16, 18, and 45 originally isolated from clinical material were provided by Professor E-M de Villiers (Referenzzentrum fur humanpathogene Papillomviren, Heidelberg, Germany) and HPV-33 was obtained from Dr G Orth (Institut Pasteur, Paris, France). HPV types 16, 18, and 33 were provided in pBR322 and HPV-45 in pGEM4. HPV-31 was cloned in pT713 and was obtained from Dr A Lorincz (Digene Diagnostics, Silver Spring, Maryland, USA).
Transformation was performed using Library Efficiency DH5α™ competent cells (Life Technologies, Paisley, Scotland, UK). Plasmid containing colonies were selected using L-Amp plates and were then cultured in L-broth containing ampicillin. Plasmid DNA was isolated from this bacterial cell culture using the Hybaid recovery quick mini spin kit (Hybaid Ltd, Middlesex, UK) according to the manufacturer's instructions. The extracted DNA was digested with selected restriction enzymes (BamHI, EcoRI, HindIII, HindII, and BglII) and electrophoresed in 1% agarose to check the identity of the DNA. Finally, GeneQuant II spectrophotometric analysis (Amersham Pharmacia Biotech, St Albans, Hertfordshire, UK) was performed to measure the concentration of DNA.
CLINICAL SAMPLES
A total of 300 liquid based cytology samples were collected from women attending general practitioner clinics for routine cervical screening or follow up. Cells were collected from the cervix using a Cervex Brush® rinsed in 20 ml of PreservCyt® (PC) solution (Cytyc Corporation; Boxborough, Massachusetts, USA). The sampler was then discarded. One hundred specimens were “split samples”, where a conventional smear had been made before the residual cervical material was rinsed into the liquid based cytology medium. The other 200 samples were collected routinely and only a ThinPrep® (TP) monolayer smear was made for diagnostic purposes.
DNA EXTRACTION
Cells from the residual volume were pelleted at 2900 ×g for 15 minutes before resuspending in 200 μl Tris EDTA (TE) buffer (pH 7.2). DNA extraction was then performed directly using the Qiagen DNA mini kit, according to the manufacturer's tissue protocol, resulting in 400 μl of extracted sample.
PRIMERS AND PCR PROTOCOL
Three primer pairs were used in our study: the GP5+/6+ consensus primer pair, together with HPV-16 and HPV-18 specific primer pairs, which were modified from GP5+/6+ (table 1). These were designed after a thorough search of the HPV sequence database (Los Alamos National Laboratory) in the GP5+/6+ primer target region of the L1 gene. The DNA sequence of all known HPV types was critically reviewed for optimal product length, annealing temperature, base variations between types, and the possibility of non-specific amplification.
Sequences of primers used and optimal cycling conditions found
Reaction mixes contained 0.5 μl of 5 μM forward and reverse primers, 1.25 μl of template nucleic acid, 0.25 μl of 1/1000 SYBR Green I, and 2.5 μl of master mix containing 4 mM MgCl2 (Biogene Ltd) and TaqStart antibody (Sigma-Aldrich Co Ltd, Poole, Dorset, UK). The cycling profiles were optimised for each set of primers and are detailed in table 1. Detailed melt analysis was used for the accurate determination of the melting point of the amplified product. This consisted of a single cycle of 65°C for three seconds to 90°C for one second at a transition rate of 0.2°C/second, followed by measurement of the fluorescent signal at greater frequency.16 The entire assay including DNA extraction, PCR amplification, and melting temperature analysis can be performed in approximately three hours.
HYBRID CAPTURE ASSAY
A 4 ml volume of PC fluid was processed for the Digene HPV hybrid capture assay (HCA) according to the manufacturer's recommendations and using the second generation (HCA II) test. This is a sandwich capture hybridisation system using chemiluminescent signal amplification for the qualitative detection of 13 different high risk HPV types (HPV types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 68). The emission of light is measured as relative light units (RLUs) and is proportional to the amount of target DNA present. Samples with an RLU > 1 were considered positive for any of the high risk HPV types contained in the probe pool. HPV results were correlated with the cytology results obtained.
CYTOLOGICAL ASSESSMENT
TP slides were made using the semi-automated ThinPrep 2000 slide processor (Cytyc Corporation, Boxborough, Massachusetts, USA). The methodology has been well documented elsewhere.17, 18 Both conventional and TP smears were reported independently. The Richart system for cervical diagnostic reporting was used (unsatisfactory (U/S), negative (WNL), borderline changes (B/L), mild dyskaryosis, moderate dyskaryosis, and severe dyskaryosis).
Results
CONTROL MATERIAL
Cultured cells containing HPV-16 or HPV-18
With GP5+/6+ primers, replicate 10-fold dilutions of SiHa cells containing 1000–2000 genome copies down to 0.1 copy of the HPV-16 genome were tested and the product gave a single peak and a sensitivity of 1–2 copies/μl. Similarly, 10-fold dilutions of HeLa cells containing 5000 down to 0.5 copies of the HPV-18 genome showed a sensitivity of 3–50 copies/μl. Detailed melt analysis of 20 samples each of SiHa and HeLa cells gave the mean Tm of the HPV-16 product as 79.1°C (2 SD, 0.8) and the mean Tm of the HPV-18 product as 80.9°C (2 SD, 1.0) (table 2). In a mixed population of SiHa and HeLa cells, a composite melting curve was observed on standard analysis, with a Tm of 79.4°C (fig 1A). On further analysis using a detailed melt cycle, this resolved into two clearly differentiated peaks with temperatures of 78.4°C and 80.1°C (fig 1B). Using this system, HPV-16 could be detected in a 1/1000 mixture of SiHa to HeLa cells.
Detailed melt analysis of DNA extracted from SiHa and HeLa cells

Melting curve analysis of PCR products after amplification of a 150 bp fragment of the human papillomavirus (HPV) L1 gene. Y axis: negative differential of fluorescence over temperature (−dF/dT). (A) Standard melt analysis of SiHa/HeLa mixed DNA product, using GP5+6+ primers showing single peak (Tm = 79.4°C). (B) Detailed melt analysis of SiHa and HeLa mixed DNA product, showing two peaks with Tm = 78.4°C (HPV-16) and 80.1°C (HPV-18).
Type specific primers, TS165+/166+ and TS185+/186+ (table 1), from a similar region of the L1 gene gave individual peaks at 79.2°C and 81.3°C for HPV-16 in SiHa and HPV-18 in HeLa cells, respectively, and detailed melt analysis of 30 samples confirmed these mean melting points as 78.2°C (2 SD, 0.4) and 80.4°C (2 SD, 0.3), respectively. The specificity of these primers led to a smaller standard deviation in Tm (table 2). Different cycling conditions were required for each set of specific primers, reflecting the optimal conditions for stringency of each pair. The differential ability of the specific primer pairs was assessed by amplifying mixtures of SiHa and HeLa cells in the presence of one or other primer pair. In mixtures with equivalent copy numbers, both types were readily detected. Again, HPV-16 from a single SiHa cell could be detected in a background of 103 HeLa cells (table 3).
Detection of human papillomavirus 16 (HPV-16) in a background of HPV-18 with GP5+/6+ and with TS 165+/166+ primers
Cloned material
Detailed melt analysis allowed the differentiation of five different types of high risk HPV from cloned material and showed a range from 76.7°C for HPV-33 to 80.4°C for HPV-18 (table 4). We are currently investigating the differentiation by Tm of additional HPV types.
Melting temperature (Tm) of high risk human papillomavirus (HPV) cloned material using real time PCR with GP5+/6+ primers and detailed melt analysis
PATIENT SAMPLES
One hundred preselected samples previously analysed using HCA and conventional PCR were assessed using real time PCR. Table 5 shows the comparison between conventional and real time PCR. Five samples were positive by real time PCR but negative by conventional PCR. Of these five discrepant samples, one contained HPV-16 as shown by use of type specific primers from both the L1 and E6 regions,15 one contained an HPV-16 related type as shown by detection with HPV-16 E6 primers but not HPV-16 L1 primers, and three contained HPV “X”, being positive by HCA but negative with HPV-16 L1 and E6 primers and also HPV-18 type specific primers.
Comparison of conventional and real time PCR on 100 selected clinical samples
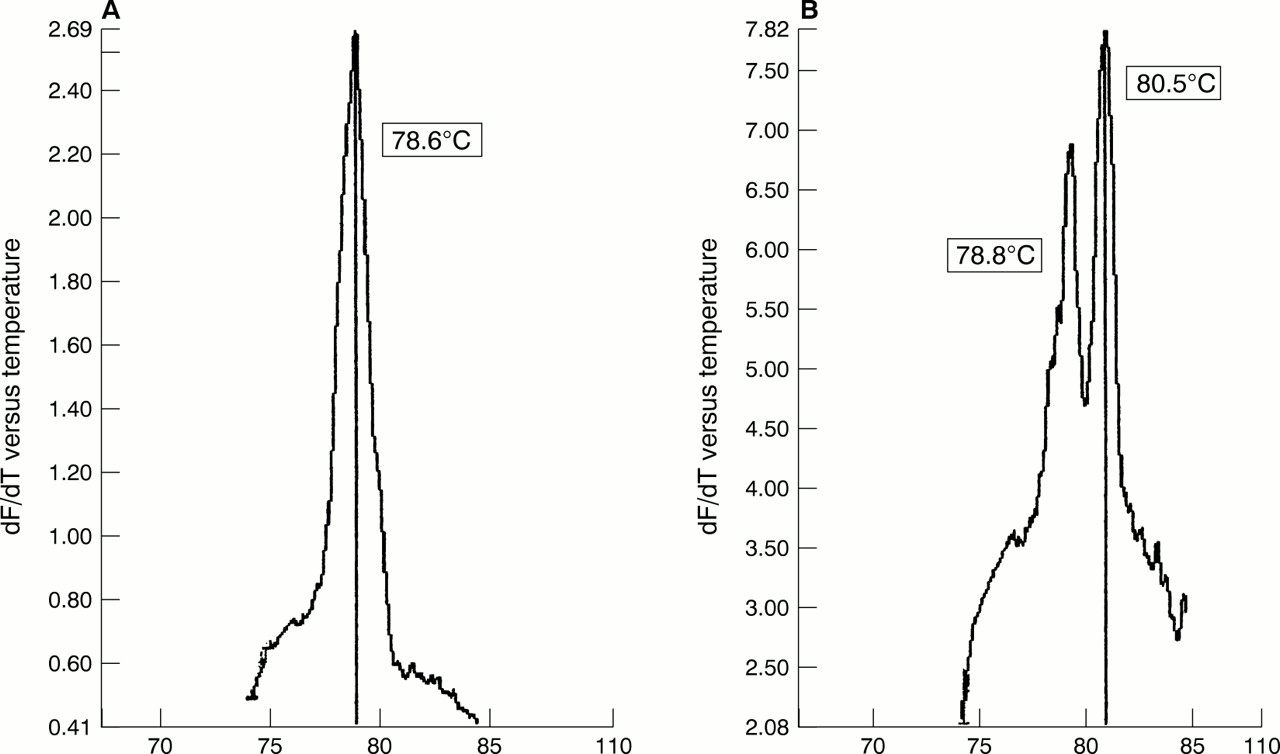
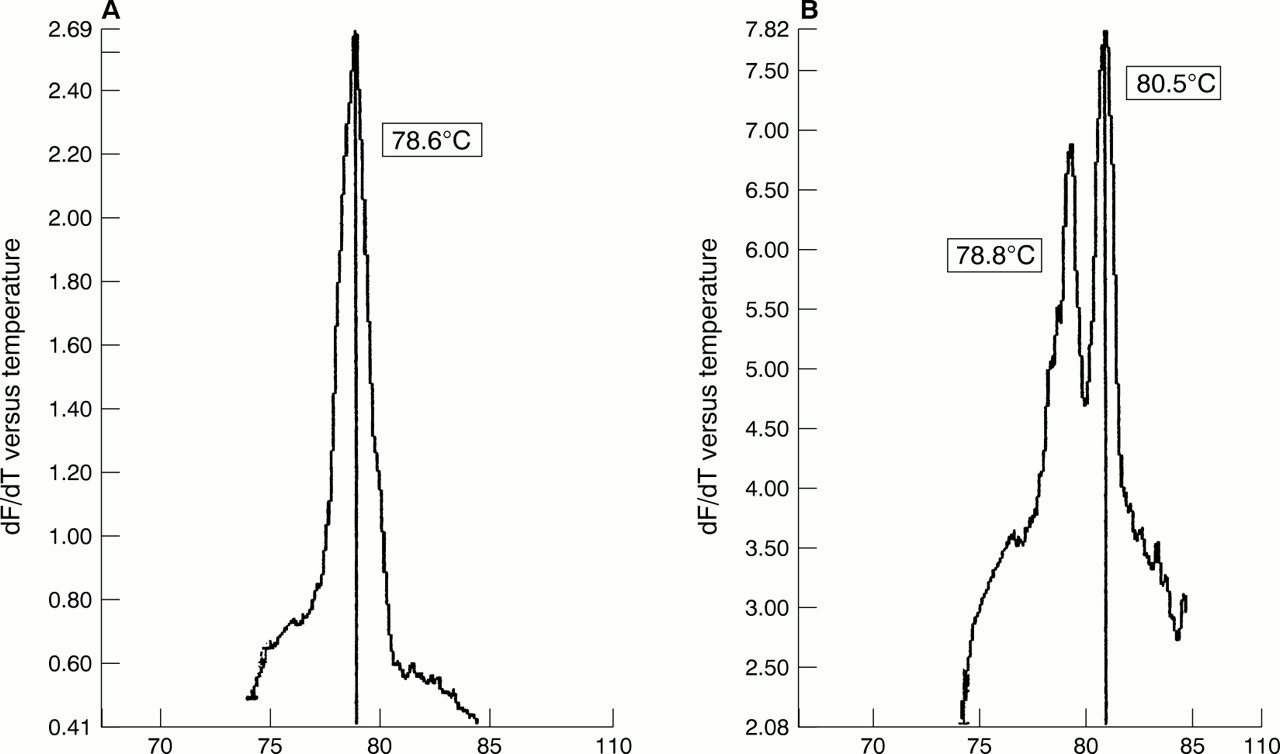
The first 200 routine cervical screening samples from our study looking at the combined effectiveness of liquid based cytology sampling and real time PCR were processed within 21 days of receipt and analysed without previous knowledge of the cytology results. Nineteen (9.5%) of the 200 samples contained HPV (table 6), of which 13 (6.8%) were found in women with no evidence of cytological abnormalities. Most of the positive samples (12 of 19) appeared to contain HPV-16 by detailed melt analysis (fig 2A) and an occasional double infection was noted (fig 2B).
Human papillomavirus (HPV) findings related to cytology in 200 unselected, routine liquid based cytology samples
{kind=link}
{kind=link}
Melting curve analysis of PCR products from clinical liquid based cytology samples. (A) Detection of a single peak of human papillomavirus 16 (HPV-16) using detailed melt analysis. (B) Detection of a mixed infection with HPV types 16 and 18 using detailed melt analysis.
Discussion
We have shown that HPV-16 and HPV-18 can be detected by rapid real time PCR using consensus primers and can be differentiated by melting curve analysis. In mixed samples, two separate peaks of distinct Tm are seen, even when there is a considerable difference in the copy number of each type. This approach has been confirmed with type specific primers.
Because HPV-16 and HPV-18 product lengths are very similar, it is impossible to differentiate between individual HPV types by agarose gel electrophoresis after GP5+/6+ PCR amplification. In contrast, melting curve analysis using LightCycler technology can distinguish between products of the same length but different GC : AT ratios.18 Woo and colleagues19 reported the use of genus specific amplification primers and specific fluorogenic hybridisation probes to differentiate pathogenic and non-pathogenic strains of leptospira. In their study, the lower limit of detection was 200 genome copies. The same group also used melting curve analysis to differentiate different strains of leptonema20 and to distinguish reference strains and field isolates of leptospira without the use of hybridisation probes.21 More recently, reports of the use of the LightCycler in detecting viruses have begun to appear. These include the quantitative detection of human cytomegalovirus (CMV) in plasma22 and the sensitive diagnosis of herpes simplex virus in clinical samples using Tm analysis to distinguish between herpes simplex virus 1 (HSV-1) and HSV-2.23 We have used a similar approach to separate HPV types, specifically for the differentiation of HPV-16 and HPV-18 products that differ in Tm by less than 2°C.
Real time PCR technology has great potential for clinical and non-clinical development. Nevertheless, it is still a new technique and some technical problems have been reported, including the presence of primer dimer formation.19 In our hands this could be minimised by careful attention to optimisation conditions, with very small changes in concentration, temperature, and the use of TaqStart antibody having a considerable effect on the shape of the analytical trace. In addition, the presence of a high molecular weight non-specific PCR product has been noted in some applications.19 Wittwer and colleagues14 suggested that “shoulders” of non-specific PCR product might be caused by substantial product to product annealing in later amplification cycles. A small shoulder was sometimes seen in our study on the high temperature side of the melting curve. The problem was minimised by reducing the amplification mix from 10 μl to 5 μl and indeed the manufacturers recommend a volume of 5–7 μl as optimal in each capillary. Larger volumes can result in uneven temperature distribution and therefore inefficient amplification towards the top portion of the capillary. With optimal conditions and volume, we only observed this phenomenon occasionally with type specific primers and it did not interfere with the determination of the Tm value. The effect was never seen with the consensus primers. Quality control of the reagents used is undoubtedly important to limit variations in Tm, and positive control material was included and fully analysed in every run.
Additional practical problems have included the fragility of the capillaries. However, breakages were minimal with experienced operators and the second generation LightCycler produced by Roche Molecular Systems uses more robust capillaries. The extended use of the LightCycler for detailed melt analysis can result in overheating of the carousel and, in our hands, only 10 detailed melt analyses could be carried out before a 30 minute cooling period was required. Nevertheless, the rapid cycling time with standard melts allowed up to seven PCR runs to be carried out in a single working day.
We used melting point analysis after amplification in a single reaction for the detection of single and mixed samples of HPV-16 and HPV-18 in both cell lines in vitro and in cervical secretions from patients, including 100 samples validated by both conventional PCR and HCA II. Subsequently, the protocol was applied successfully to 200 liquid based cytology clinical samples, with HPV DNA being detected in 9.8% of samples (table 6), including 13 of 189 (6.8%) showing no cytological abnormality. Although this is consistent with other studies,11, 24 analysis of the HPV results in relation to cervical dyskaryosis requires a much larger study group and this work is currently under way. The combined approach of rapid amplification and product identification in a single PCR reaction is an exciting one, with great potential for both clinical and non-clinical development, particularly in terms of introducing HPV testing into cervical screening programmes. Opportunities for high throughput are possible by combining one of several available robotic handling instruments for DNA extraction with the LightCycler system and we are testing the maximum daily capacity of such combinations.
Acknowledgments
We are grateful to Professor E-M de Villiers (Referenzzentrum fur humanpathogene Papillomviren, Heidelberg, Germany) for providing HPV types 16, 18, and 45; Dr G Orth (Institut Pasteur, Paris, France) for HPV-33; and Dr A Lorincz (Digene Diagnostics, Silver Spring, Maryland, USA) for HPV-31. We acknowledge the help of Dr B Morris, department of pathology, University of Edinburgh with growth and isolation of the plasmid DNAs. We would like to thank the Chief Scientist Office of the Scottish Executive for funding this work (Grant No K/MRS/50/C2699) and the NHS R&D Support Fund for additional support.