Article Text

Abstract
Aims—To ascertain the clinical relevance of a strain of Enterobacteriaceae isolated from the stool of a bone marrow transplant recipient with diarrhoea. The isolate could not be identified to the genus level by conventional phenotypic methods and required 16S ribosomal RNA (rRNA) gene sequencing for full identification.
Methods—The isolate was investigated phenotypically by standard biochemical methods using conventional biochemical tests and two commercially available systems, the Vitek (GNI+) and API (20E) systems. Genotypically, the 16S bacterial rRNA gene was amplified by the polymerase chain reaction (PCR) and sequenced. The sequence of the PCR product was compared with known 16S rRNA gene sequences in the GenBank database by multiple sequence alignment.
Results—Conventional biochemical tests did not reveal a pattern resembling any known member of the Enterobacteriaceae family. The isolate was identified as Salmonella arizonae (73%) and Escherichia coli (76%) by the Vitek (GNI+) and API (20E) systems, respectively. 16S rRNA sequencing showed that there was only one base difference between the isolate and E coli K-12, but 48 and 47 base differences between the isolate and S typhimurium (NCTC 8391) and S typhi (St111), respectively, showing that it was an E coli strain. The patient did not require any specific treatment and the diarrhoea subsided spontaneously.
Conclusions—16S rRNA gene sequencing was useful in ascertaining the clinical relevance of the strain of Enterobacteriaceae isolated from the stool of the bone marrow transplant recipient with diarrhoea.
- 16S ribosomal RNA sequencing
- bone marrow transplantation
Statistics from Altmetric.com
The identification of bacteria in the clinical microbiology laboratory is performed traditionally by isolating the organism and studying it phenotypically by means of Gram staining, culture, and biochemical methods, which have been the gold standard of bacterial identification. However, these methods of bacterial identification have two major drawbacks. First, they cannot be used for non-cultivable organisms such as Tropheryma whippelii. Second, we are occasionally faced with organisms with biochemical characteristics that do not fit into patterns of any known genus and species.
Since the discovery of the polymerase chain reaction (PCR) and DNA sequencing, the genomes of some bacteria have been sequenced completely.1 A comparison of the genomic sequences of bacterial species showed that the 16S ribosomal RNA (rRNA) gene is highly conserved within a species and among species of the same genus, and hence can be used as the new gold standard for the speciation of bacteria. Using this new standard, phylogenetic trees based on base differences between species are constructed; bacteria are classified and re-classified into new genera;2,3 and classifications of non-cultivable microorganisms are made possible.4,5 It can also be useful in elucidating the relation of unknown bacterial species to known ones, and new species of bacteria such as Gemella sanguinis, Mycobacterium heidelbergense, and Massilis timonae have been discovered using 16S rRNA sequencing.6–8 Moreover, bacteria such as Mycobacterium celatum and Methylobacterium zatmanii, which were not known to cause infections in humans have been identified in clinical specimens using this technique.9,10 Furthermore, bacteria that are difficult to identify were speciated successfully using this technique.11 In our study, we report the application of such a technique to ascertain the clinical relevance of a strain of Enterobacteriaceae isolated from the stool of a bone marrow transplant recipient with diarrhoea.
Methods
PATIENT, SPECIMEN COLLECTION, AND MICROBIOLOGICAL METHODS
Specimen collection and microbiological methods were described in a previously published paper.12 Briefly, routine surveillance cultures of throat and rectal swabs were performed weekly in the first 30 days after bone marrow transplantation, and when diarrhoea occurred, stool was collected and cultured for potential pathogens. All suspect colonies were identified by standard conventional biochemical methods.13 In addition, the Vitek System (bioMerieux Vitek, Hazelwood, Missouri, USA) and the API system (bioMerieux Vitek) were used for the identification of the bacterial isolate in our study.
EXTRACTION OF BACTERIAL DNA FOR 16S RIBOSOMAL RNA GENE SEQUENCING
Bacterial DNA extraction was modified from a published protocol.14 An aliquot of 80 μl of NaOH (0.05 M) was added to 20 μl of bacterial cells suspended in distilled water and the mixture was incubated at 60°C for 45 minutes, followed by the addition of 6 μl of Tris/HCl (pH 7.0), achieving a final pH of 8.0. The resultant mixture was diluted 100 times and 5 μl of the diluted extract was used for PCR.
PCR, GEL ELECTROPHORESIS, AND 16S RIBOSOMAL RNA GENE SEQUENCING
PCR amplification of the 16S rRNA gene was modified from a published protocol.15 DNase I treated distilled water and PCR master mix (which contains deoxynucleoside triphosphates (dNTPs), PCR buffer, and Taq polymerase) were used in all PCR reactions by adding 1 U of DNase I (Pharmacia, Uppsala, Sweden) to 40 μl of distilled water or PCR master mix, incubating the mixture at 25°C for 15 minutes, and subsequently at 95°C for 10 minutes to inactivate the DNase I. The bacterial DNA extract and control were amplified with 0.5 μM primers (LPW57, 5′-AGTTT GATCCTGGCTCAG-3′; and LPW58, 5′-AGGCCCGGGAACGTATTCAC-3′) (Gibco BRL, Rockville, Maryland, USA). The PCR mixture (50 μl) contained bacterial DNA, PCR buffer (10 mM Tris/HCl (pH 8.3), 50 mM KCl, 3 mM MgCl2, and 0.01% gelatin), 200 μM of each dNTP, and 1.0 U Taq polymerase (Boehringer Mannheim, Mannheim, Germany). The mixtures were amplified for 40 cycles at 94°C for one minute, 55°C for one minute, and 72°C for two minutes, with a final extension at 72°C for 10 minutes in an automated thermal cycler (Perkin-Elmer Cetus, Gouda, The Netherlands). DNase I treated distilled water was used as the negative control. An aliquot of 10 μl of each amplified product was electrophoresed in 1.0% (wt/vol) agarose gel, with a molecular size marker (SPP1 EcoRI digest; Boehringer Mannheim) in parallel. Electrophoresis in Tris-borate-EDTA buffer was performed at 100 V for 1.5 hours. The gel was stained with ethidium bromide (0.5 μg/ml) for 15 minutes, rinsed, and photographed under ultraviolet light illumination.
The PCR product was gel purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany). Both strands of the PCR product were sequenced twice with an ABI 310 automated sequencer according to manufacturers' instructions (Perkin-Elmer, Foster City, California, USA), using the PCR primers (LPW57 and LPW58) and additional primers designed from the sequencing data of the first round of the sequencing reaction (LPW69 and LPW70; table 1). The sequence of the PCR product was compared with known 16S rRNA gene sequences in the GenBank database by multiple sequence alignment using the CLUSTAL W program.16
Oligonucleotides used for sequencing the 16S ribosomal RNA gene
Results
PATIENT
The 31 year old patient was diagnosed with acute myeloid leukaemia (M2) in March 1998. The disease was in first complete remission after chemotherapy. She received a syngeneic bone marrow transplant in July 1998 after conditioning with busulfan/cyclophosphamide. Three days before the bone marrow transplant she developed diarrhoea. A strain of Enterobacteriaceae that produced hydrogen sulphide and agglutinated with poly O and poly H salmonella antisera was isolated from the patient's stool on deoxycholate citrate agar and xylose lysine deoxycholate agar. The strain was persistently recovered from the rectal swabs taken in the first and third weeks after her bone marrow transplant for surveillance culture, but not in any other specimens. The marrow engrafted on day 14 and the bone marrow transplant was uneventful. She was discharged on day 24.
IDENTIFICATION OF THE BACTERIAL STRAIN BY CONVENTIONAL METHODS AND COMMERCIALLY AVAILABLE SYSTEMS
The bacterial strain was a Gram negative, facultative anaerobic rod. It grew on blood agar, chocolate agar, and MacConkey agar to sizes of 4 mm in diameter after 24 hours of incubation at 37°C in ambient air. It fermented glucose, reduced nitrate, and did not produce cytochrome oxidase, typically a member of the Enterobacteriaceae family. Standard conventional or commercially available biochemical tests did not reveal a pattern resembling any known member of the Enterobacteriaceae family (table 2). The Vitek system (GNI+) showed that it was 73% Salmonella arizonae and 17% Salmonella spp; whereas the API system (20E) showed that it was 76% Escherichia coli and 23% S arizonae. In addition, the isolate was motile, and it agglutinated with poly O and poly H salmonella antisera (Murex Biotech Ltd, Temple Hill, Dartford, UK), but did not agglutinate with any individual O, H, or Vi salmonella antisera. The strain was resistant to ampicillin, cephalothin, cefuroxime, gentamicin, cotrimoxazole, and ciprofloxacin; but sensitive to cefoxitin, ceftriaxone, amoxicillin/clavulanic acid, ceftazidime, amikacin, and imipenem.
Biochemical profile and identification of the isolate from the BMT recipient by conventional biochemical tests, the Vitek GNI+ system, and the API 20E system
PCR AMPLIFICATION AND 16S RIBOSOMAL RNA GENE SEQUENCING
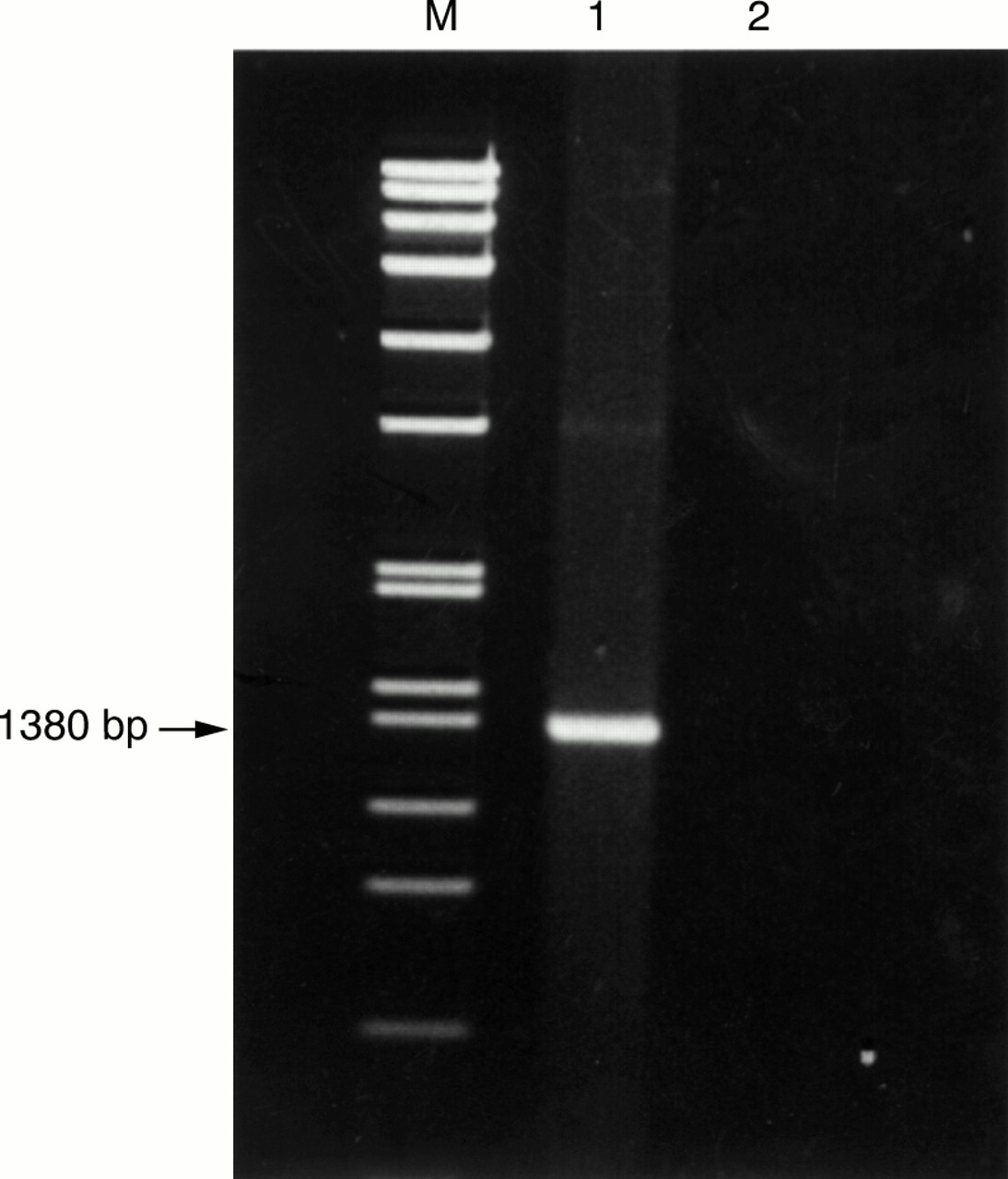
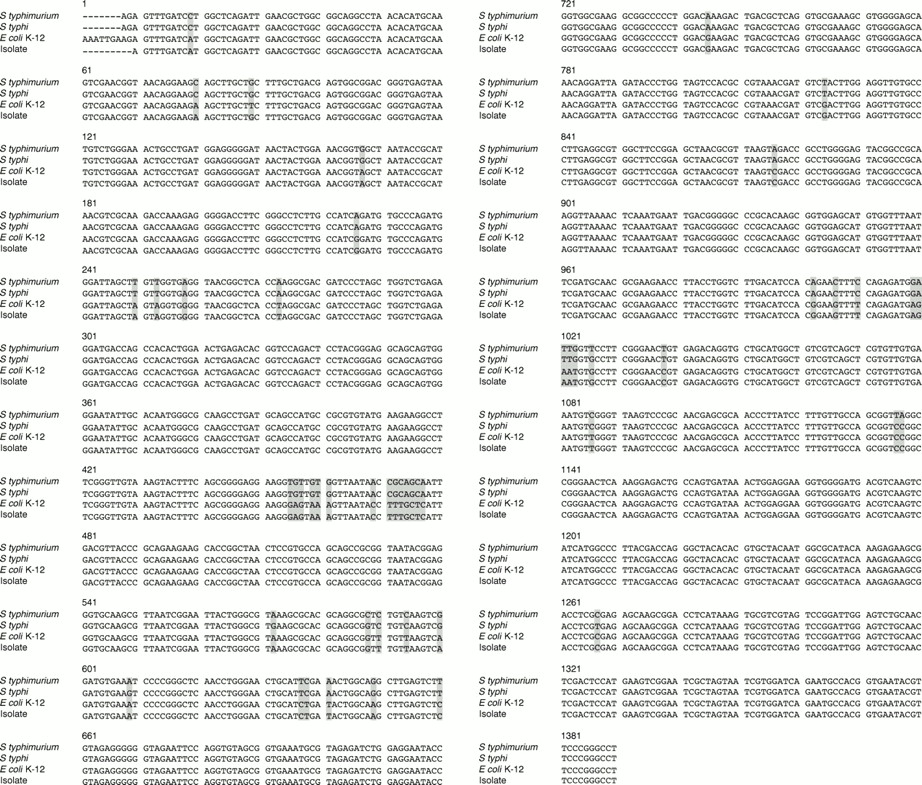
PCR of the 16S rRNA gene of the bacteria showed a band at 1380 bp (fig 1). Figure 2 shows the base sequences of the purified band and the corresponding region in E coli K-12, S typhimurium, and S typhi. There was only one base difference between the isolate and E coli K-12, but 48 and 47 base differences between the isolate and S typhimurium (NCTC 8391) and S typhi (St111), respectively, showing that the isolate was a strain of E coli.

DNA products from PCR of 16S ribosomal gene. Lane M, molecular marker SPP1 EcoRI digest; lane 1, bacterial isolate from bone marrow transplant recipient; lane 2, negative control containing DNase I treated distilled water.

{kind=link}
{kind=link}
DNA sequences of the 16S rRNA gene of the isolate from the bone marrow transplant recipient, Escherichia coli K-12, Salmonella typhimurium (NCTC 8391), and Salmonella typhi (St111). The shaded bases represent those in the isolate that are different from the corresponding ones in E coli K-12, S typhimurium (NCTC 8391), or S typhi (St111).
Discussion
Immunocompromised hosts have always been one of the most important sources of emerging pathogens and novel antimicrobial resistance. Vancomycin resistant Staphylococcus aureus strains have been found in patients with leukaemia, lung cancer, stomach cancer, mycosis fungoides, chronic renal failure, and diabetes mellitus17,18; and multiresistant species of pseudomonas and acinetobacter are usually found in immunocompromised patients in intensive care units.19 Bartonella and human herpesvirus 8 were important emerging and re-emerging pathogens in patients infected with human immunodeficiency virus. Emerging pathogens, such as Ochrobactrum intermedium and Legionella parisiensis, have been isolated and identified using 16S rRNA sequencing in liver transplant recipients.20,21 Bone marrow transplantation is a situation where profound suppression of both the innate and adaptive immunity occurs, and the gastrointestinal tracts of bone marrow transplant recipients contain numerous species of microorganisms. Cytotoxic agents and other selective pressures will lead to a high frequency of induced mutations in the microorganisms in the gastrointestinal tract of bone marrow transplant recipients and acquisition of genes from other microorganisms in the surroundings that can result in a survival advantage for the bacteria.
We describe a strain of the Enterobacteriaceae family isolated from the gastrointestinal tract of a bone marrow transplant recipient. Conventional biochemical tests failed to differentiate between species of E coli and salmonella. Reactions not favouring salmonella included metabolism of orthonitrophenyl galactoside (ONPG) (2%) and production of indole (1%). The only ONPG positive salmonellae are S arizonae, S diarizonae, S bongori, and S indica, but the isolate was malonate positive (which was not in favour of S bongori and S indica), gelatinase negative (which was not in favour of S arizonae and S diarizonae), and did not agglutinate with any “individual” O or H salmonella antisera. Furthermore, none of the four salmonella species produces indole. Reactions not favouring E coli include the production of hydrogen sulphide and the agglutination of poly O and poly H salmonella antisera. Serotyping for EPEC and the Sereny test for ETEC were negative. Furthermore, although Citrobacter freundii is famous for crossreacting with poly O and poly H antisera, reactions that preclude it include citrate utilisation, production of indole, and production of lysine decarboxylase.
The identification of the organism in our study was important because the management of the patient would be radically different depending on which organism was identified. If the organism is a strain of E coli (as it was in our patient) no treatment is needed. This is because of evidence that, despite persistent recovery of the bacteria in routine surveillance cultures in the first and third weeks after bone marrow transplantation, the patient's diarrhoea will subside spontaneously without any specific treatment. On the other hand, if the organism turns out to be an ampicillin, cotrimoxazole, and ciprofloxacin resistant species of salmonella, ceftriaxone would have to be administered to the patient to prevent the development of infection in the gastrointestinal tract or invasion into the systemic circulation.
16S rRNA sequencing will continue to be the gold standard for bacterial identification. With the help of additional tests, the Vitek GNI+ and API 20E systems were only able to identify 80.1–94.4% and 95.6–98.6%, respectively, of Enterobacteriaceae and common non-glucose fermenting Gram negative bacilli.22–24 Inert E coli are notoriously difficult to identify, and in one report, two atypical S enteritidis strains were misidentified by both systems.23 Whenever the identification of an organism is crucial, as in this case, 16S rRNA sequencing should be carried out for these “difficult to identify” strains. This is in line with a recently published report, showing that a technique using 16S rRNA sequencing was able to identify 97.2% of 72 unusual aerobic Gram negative bacilli, which was significantly better than methods based on cellular fatty acid profiles (77.8%) or carbon source utilisation (87.5%).25
The use of DNA chip technology might enable 16S rRNA sequencing to be used routinely in clinical microbiology laboratories, replacing the traditional biochemical tests. Manpower, and hence money, have been saved by an increase in the use of automation in clinical microbiology laboratories. Such technology has been used for the detection of bacterial growth in blood culture bottles and urine samples, the measurement of antibiotic concentrations and antibiotic resistance, and pipetting of serum samples for various kinds of serological tests. Modern technologies have made it possible to construct a high density of oligonucleotide arrays on a chip with oligonucleotides representing the 16S rRNA sequence of bacteria. Such a design will facilitate automation of the annealing process and detection of the PCR products of 16S rRNA amplification, hence making the identification of clinical isolates possible. Although the cost effectiveness of using 16S rRNA sequencing in routine clinical microbiology laboratories remains to be evaluated, the present example has shown the usefulness of 16S rRNA sequencing for ascertaining the relevance of a clinical isolate.
Acknowledgments
This work was partly supported by the committee of research and conference grants, the University of Hong Kong. We thank Dr R Lee for comments on the manuscript.