Article Text

Abstract
The type 1 insulin-like growth factor receptor (IGF-IR) sends several signals, some of which are contradictory. When the concentrations of insulin receptor substrate 1 (IRS-1), a major substrate of the IGF-IR, are high, the signal is mitogenic, anti-apoptotic, and can even cause malignant transformation. However, in the absence of IRS-1, the IGF-IR sends a differentiation signal, which leads to granulocytic differentiation in haemopoietic cells. The mitogenic signal of the IGF-IR/IRS-1 combination depends largely, but not exclusively, on the activation of the phosphatidylinositol-3 kinase (PI3K).
- insulin-like growth factor 1 receptor
- differentiation
- insulin receptor substrate 1
- transformation
Statistics from Altmetric.com
The type 1 insulin-like growth factor receptor (IGF-IR), activated by its ligands, controls the proliferation of cells in a variety of ways, namely: (1) It sends a mitogenic signal. (2) It protects cells from a variety of apoptotic injuries. (3) It promotes growth in cell size (a requirement for cell division). (4) It plays a crucial role in the establishment and maintenance of the transformed phenotype. (5) It regulates cell adhesion and cell motility. (6) It can induce terminal differentiation. The IGF-IR is expressed in most cell types, the only known exceptions being hepatocytes and mature B cells. Because the IGF-IR is found ubiquitously, it participates in the growth regulation of many cell types, even though the primary growth factor might be another molecule. These various functions of the IGF-IR have been discussed in recent reviews, to which the reader is referred for detailed references.1–5 In our present communication, we wish to focus on two characteristics of the IGF-IR that are seemingly contradictory; that is, the ability of the receptor to send signals for either malignant transformation or differentiation.
Although the mitogenicity of the IGF-IR has been known for a long time,6 there is also a substantial amount of literature on its ability to send a differentiation signal (reviewed in Baserga and Morrione3). For instance, under certain conditions, myoblasts, osteoblasts, adipocytes, oligodendrocytes, neurones, and haemopoietic cells can be induced to differentiate by IGF-I or IGF-II. The role of the IGF system in differentiation and transformation can be studied in haemopoietic cells. Differentiation of haemopoietic cells occurs only if the cells first undergo one or two rounds of replication.7–9 This is true for instance for the granulocytic colony stimulating factor (G-CSF), as well as for IGF-I mediated differentiation.10 Initially, we will review the role of the IGF-IR in malignant transformation and in differentiation. Because IRS-1 plays a dominant role in directing IGF-IR signalling towards either transformation or differentiation, we will later examine additional, new data on the role of IRS-1 in these two processes.
The IGF-IR and the transformation of cells
The role of the IGF-1R in malignant transformation rests on two fundamental observations. The first one is the singular resistance to transformation of mouse embryo fibroblasts with a targeted disruption of the IGF-IR genes.11 The second observation is that the downregulation of IGF-IR function causes massive apoptosis in tumour cells growing in anchorage independent conditions.2 Mouse embryo fibroblasts with a disruption of the IGF-IR genes,12, 13 called R− cells, have been found to be resistant to transformation by a variety of viral and cellular oncogenes. The list of oncogenes that fail to transform R− cells include the SV40 T antigen and/or an activated Ha-ras oncogene,11, 14 the bovine papillomavirus E5 protein,15 the human papillomavirus E7 protein,16 the Ewing's sarcoma fusion protein,17 an activated c-src,18 and others.5, 19, 20 All the agents that failed to transform R− cells readily transformed mouse embryo fibroblasts with endogenous IGF-IR. Accordingly, re-introduction of an IGF-IR into R− cells promptly renders these cells susceptible to transformation. To date, the only oncogenes known to transform R− cells are v-src18 and a mutant of Gα13.21
This observation is quite remarkable, because mouse embryo fibroblasts—for example, 3T3 cells from several strains of mice—have a very strong tendency to transform spontaneously in culture (reduced growth factor requirements, foci formation in monolayer cultures, and formation of colonies in soft agar). An overexpressed IGF-IR is itself transforming,22–24 but this is of doubtful meaning because almost anything overexpressed in mouse embryo fibroblasts is transforming. In the case of R− cells, our interpretation is that there is a signal originating from the IGF-IR that facilitates and is “quasi-necessary” for transformation by the usual agents (physical, chemical, genetic, etc). For instance, the IGF-IR could downregulate the expression of a tumour suppressor gene. This tumour suppressor gene could be strongly expressed in cells devoid of the IGF-IR, resulting in the suppression of transformation.3
From the time of our original observation that the SV40 T antigen failed to transform R cells11 we hypothesised as a corollary that the downregulation of the IGF-IR in malignant cells ought to reverse the transformed phenotype. In fact, downregulation of IGF-IR function, either by antisense strategies or by dominant negative mutants, causes massive apoptosis of tumour cells in vitro and in vivo. As a consequence of the extensive apoptosis of tumour cells, downregulation of IGF-IR function results in inhibition of tumorigenesis and metastases in experimental animals. The evidence and the references on this topic have been summarised in a recent review by Baserga.5 An interesting feature of IGF-IR targeting is that it has only a modest effect on cells in monolayer cultures. It seems that the IGF-IR is not an absolute requirement for normal growth, as confirmed also by genetic experiments.25 It is, however, a strict requirement for anchorage independent growth.26 Human prostatic tumour cells stably expressing a dominant negative mutant of the IGF-IR are not inhibited when growing in monolayer, but fail to form colonies in soft agar or tumours in mice.27 This differential effect on normal growth (cells in monolayer cultures) and abnormal growth (anchorage independent growth) indicates that targeting of the IGF-IR is a promising candidate for use in cancer chemotherapy.
The IGF-IR in differentiation
As already mentioned, the response of haemopoietic cells to IGF-I can be studied in 32D cells. 32D cells are murine haemopoietic cells of the myeloid lineage, which undergo apoptosis within 24 hours of withdrawal of interleukin 3 (IL-3).7 32D cells have very low levels of IGF-IR and do not express IRS-1 or IRS-2,28, 29 which are among the major substrates of the IGF-IR and the insulin receptor (IR).30 When 32D cells express a human IGF-IR cDNA (32D IGF-IR cells), they survive in the absence of IL-3 and, with the addition of IGF-I, they grow exponentially for about 48 hours.31 After 48 hours, the cells begin to differentiate along the granulocytic pathway, and eventually decrease in number,31 as one would expect from terminally differentiated cells. If 32D IGF-IR cells are stably transfected with IRS-1 cDNA, to generate 32D IGF-IR/IRS1 cells, the cells no longer differentiate,31 grow indefinitely in the absence of IL-3, and form tumours in animals.32 It should be mentioned that 32D cells expressing only the IGF-IR cannot be passaged without IL-3, and neither do they form tumours in animals. 32D cells overexpressing only IRS-1 do not even survive for 24 hours in the absence of IL-3.29, 31, 33 Thus, individually, IRS-1 or the IGF-IR cannot transform 32D cells, or even induce their prolonged survival. However, in combination, they cause malignant transformation of 32D cells.
An intriguing aspect of this model (32D/IGF-IR cells versus 32D/IGF-IR/IRS-1 cells) is that 32D IGF-IR cells are stimulated to proliferate for the first 48 hours, before they differentiate. It means that, in terms of cell proliferation, one could not detect any difference for the first two days between 32D IGF-IR and 32D IGF-IR/IRS-1 cells. Although both cell lines are implementing a proliferative programme, only 32D IGF-IR cells are also initiating a differentiation programme. This was demonstrated by looking at an early marker of differentiation—myeloperoxidase (MPO) mRNA. In 32D IGF-R cells, MPO mRNA is already detectable within 24 hours of changing the cells from IL-3 to IGF-I. No changes in MPO RNA values occur in 32D IGF-IR/IRS1 cells.32 Thus, it seems that the IGF-IR does indeed activate both a proliferative and a differentiation programme, with the differentiation programme eventually prevailing. The expression of IRS-1 extinguishes the differentiation programme, leaving the proliferative programme intact.
Regulation of cell size and the activation of p70S6K
One of the pathways that must be activated by the IGF-IR to send a mitogenic signal is through IRS-1, a substrate of the IGF-IR, which is a potent activator of phosphatidylinositol-3 kinase (PI3K).30, 34 IRS-1 sends a strong mitogenic signal and, when overexpressed, transforms cells.35, 36 The IRS-1–PI3K pathway results in the activation of p70S6K,37 a kinase that plays an important role in cell proliferation.38 The IRS-1-mediated inhibition of cell differentiation in 32D IGF-IR cells involves a sustained activation of p70S6K.32 In fact, an inhibitor of p70S6K activity, rapamycin, causes the differentiation of 32D IGF-IR/IRS-1 cells.32 In addition, ectopic expression of IRS-1 causes an increase in the size of 32D IGF-IR cells.32 This is in agreement with results obtained in drosophila and mice. The homologues of IRS-1,39 Akt and p70S6K,40, 41 regulate the size of drosophila, and disruption of the p70S6K gene in mice results in a small mouse phenotype.42 In turn, the effect of this pathway on cell and animal size might be related to the ability of the activated IGF-IR to regulate, directly or indirectly, the synthesis of ribosomal RNA.43–45
Mechanism(s) of IRS-1 inhibition of differentiation
The crucial role of IRS-1 in inhibiting differentiation and causing malignant transformation has prompted us to investigate the mechanism(s) involved in this remarkable switching of programmes. As a beginning, we have generated several mutants of IRS-1 and have asked which domains of IRS-1 are necessary for the inhibition of differentiation. The results of this inquiry constitute the second part of our communication, which presents new data. Figure 1A shows the IRS-1 mutants that we have used and fig 1B gives their levels of expression. All cell lines were mixed populations obtained by the transduction of 32D IGF-IR cells with a retroviral vector,46 expressing the appropriate construct. The mutants, generated by polymerase chain reaction (PCR) mutagenesis46 included a deletion of the PTB domain (δPTB), a deletion of the Pleckstrin domain (δPH), and point mutants at Y608, Y891, and Y935, single or multiple. Several of these mutant IRS-1 molecules have also been studied in 32D cells by Yenush et al,47 but there are two important differences between their studies and ours. Yenush and colleagues47 used the IR, which, by itself, cannot protect 32D cells from apoptosis,48 even for 24 hours. Furthermore, Yenush and colleagues47 limited their studies to short term events (first 48 hours), whereas our studies focus on long term events, such as transformation and differentiation. Nonetheless, their experiments have been extremely useful in planning our approach.

Diagram of the mutations in insulin receptor substrate 1 (IRS-1) and the expression of IRS-1 mutants. (A) diagram of IRS-1 and the relevant domains and residues that were mutated (see text). IRS-1 mutations were generated by site directed mutagenesis as described in detail by Romano et al.46 (B) Western blot of lysates from mixed populations of cells transduced with wild-type or mutant IRS-1 expressing plasmids. Below is a western blot showing the degree of expression of the type 1 insulin-like growth factor receptor (IGF-IR) in the same cell lines. The upper band is the proreceptor. The antibodies used to visualise IRS-1 or the IGF-IR are those given in Valentinis et al.31
Figure 2 summarises the results on growth and differentiation of these mixed populations expressing mutant IRS-1 proteins. All these cell lines grew very well in IL-3 and all underwent apoptosis after IL-3 withdrawal, unless supplemented with IGF-I. Figure 2A shows the growth of these cell lines in IGF-I, at four and six days after IL-3 withdrawal. Because all of these cell lines were generated from 32D IGF-IR cells, they all grew in the first 48 hours, then two of the cell lines stopped growing and even decreased in number. The two cell lines are those transfected with an empty retroviral vector (therefore similar to parental 32D IGF-IR cells), or with the δPTB mutant of IRS-1. The growth of the cells with mutations at tyrosines 608 and 935 (binding sites for the p85 subunit of PI3K) was somewhat slower, but they kept growing from days 4–6. Aliquots of these cells were taken at day 4 and re-plated under the same condition for another four days, to test their IL-3 independence. The cell lines that grew in the first six days grew again in the absence of IL-3 for another four days, thus showing that they had acquired IL-3 independence. The empty vector and the δPTB cell lines again failed to grow.
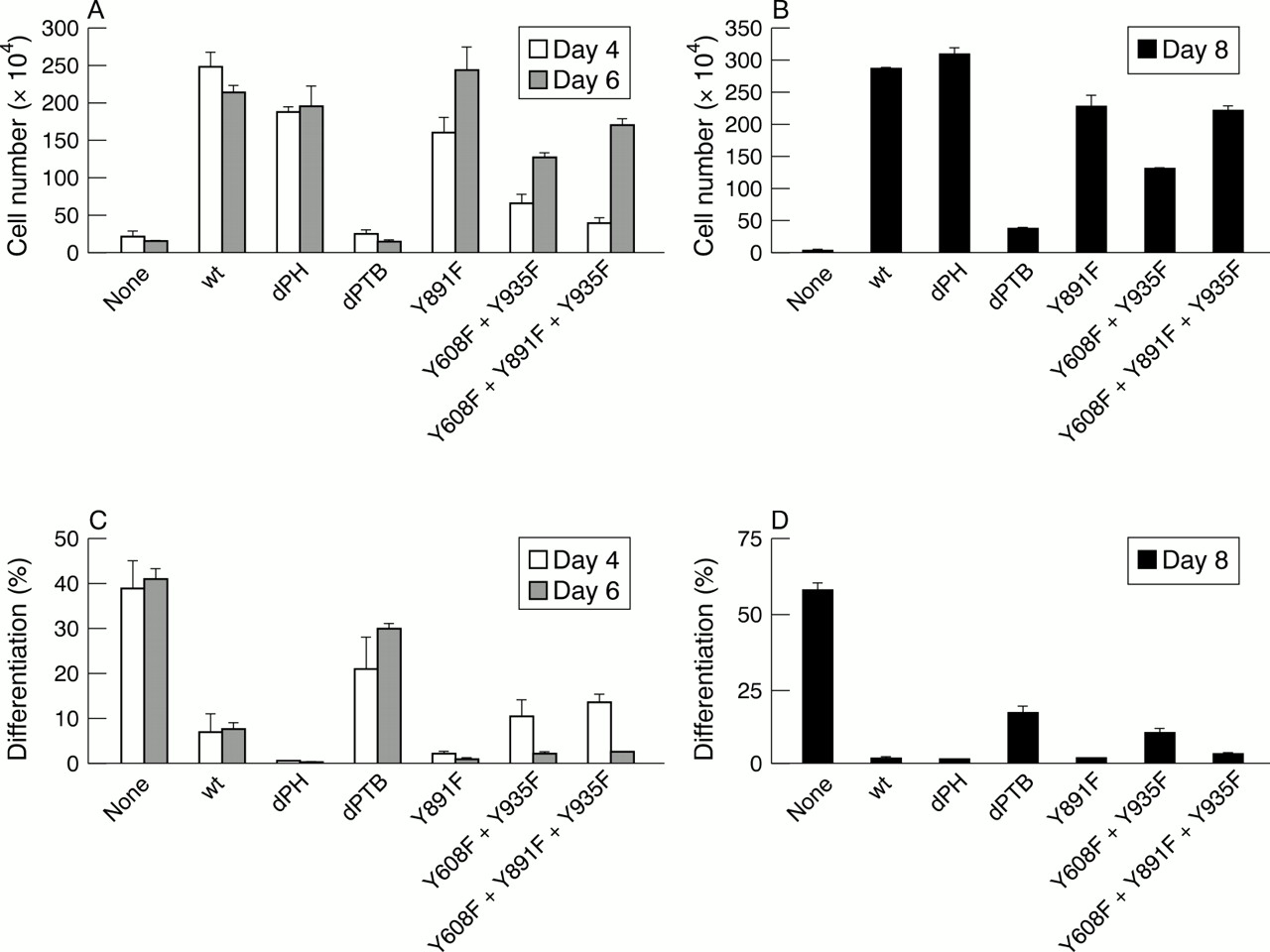
Growth and differentiation of 32D type 1 insulin-like growth factor receptor (IGF-IR) cells expressing wild-type IRS-1 or its mutants. (A) Cell number of various mixed populations at four (open bars) and six (closed bars) days after interleukin 3 (IL-3) withdrawal and supplementation with IGF-I. The cell lines are indicated on the abscissa. (B) After four days in IL-3 deficient medium, equal numbers of cells from each mixed population were re-plated and tested for their ability to grow again in the presence of IGF-I. (C) Percentage of differentiated cells in the same cell lines as in (A), at four (open bars) and six (closed bars) days after IL-3 withdrawal and the addition of IGF-1. (D) Differentiation of cells from the same mixed populations, after re-plating as in (B). The methods used to determine growth and differentiation are described in Valentinis et al.31
Figure 2C and D shows the extent of differentiation of the same mixed populations under the same conditions. These two panels can be summarised by saying that growth and differentiation are inversely correlated, as one would expect. The less the cells grow, the more they differentiate. These experiments were repeated several times by different operators, with consistent results. They show that the PTB domain of IRS-1 is necessary for the inhibition of differentiation. It can be argued that differentiation in 32D IGF-IR/δPTB cells is somewhat inhibited, when compared with the parental 32D IGF-IR cells. Nonetheless, the extent of differentiation is substantial and clearly above that of cells expressing the wild-type IRS-1 or other mutant IRS-1 molecules.
Activation of p70S6K correlates with growth
As already mentioned, the inhibition of IGF-I mediated differentiation by ectopic expression of IRS-1 correlates with the sustained activation of p70S6K.32 We determined p70S6K activation by using an antibody to phosphorylated threonine 389.32 Figure 3 shows the results of a typical experiment. All the mutant IRS-1 molecules that were active in promoting growth also gave a sustained p70S6K activation, extending to at least 60 minutes after IGF-I stimulation. As already reported, p70S6K activation is reduced in 32D IGF-IR cells (no IRS-1). It is also reduced in 32D IGF-IR cells, expressing the δPTB mutant of IRS-1.

{kind=link}
{kind=link}
{kind=link}
Activation of p70S6K in 32D type 1 insulin-like growth factor receptor (IGF-IR) cells expressing wild-type or mutant IRS-1. (A) And (C) are western blots of lysates from the indicated cell lines, using an antibody against phosphothreonine 389. The times after IGF-I stimulation are indicated above the lanes (in minutes). (B) And (D) are the same blots, stripped and re-probed with an antibody to p70S6K to monitor the amounts of protein in each lane. The methods and antibodies used were described in Valentinis et al.32
These results confirm and extend the observations of Valentinis et al,31 indicating the ability of IRS-1 to inhibit the IGF-I mediated differentiation of 32D IGF-IR cells. If IRS-1 signalling is disabled by a deletion of the PTB domain,47 differentiation is not inhibited. We did not test all the cell lines for growth in nude mice, but we tested the 32D IGF-IR cells expressing the (active) IRS-1 mutant with mutations at the p85 binding sites. Although these cells grew more slowly in nude mice than did 32D IGF-IR/IRS1 cells, they did form tumours (data not shown). We confirm also that the action of IRS-1 correlates with the sustained activation of p70S6K.32
The picture that is emerging is the dual signalling of the IGF-IR, one signal inducing proliferation, and a simultaneous signal inducing differentiation. In the absence of functional IRS-1, the differentiation signal eventually prevails over the proliferation signal, as happens also with other growth factors that induce the differentiation of haemopoietic or neuronal cells. Expression of IRS-1 (or one of its active mutants) extinguishes the differentiation programme, leaving the proliferative programme intact. There are two additional comments that should be made. The first is that IRS-1 exerts a profound negative influence on differentiation. Cells prone to differentiation often do not express IRS-1, or express very small amounts.49 Furthermore, stimuli that induce differentiation often cause a decrease in IRS-1.50 Conversely, overexpression of IRS-1 consistently decreases the extent of differentiation.31, 32 The second comment concerns the pathways used by the IGF-IR for mitogenesis and survival. It is generally agreed that the main mitogenic/survival pathway depends on the activation of the PI3K pathway.1 However, Peruzzi et al have found that, in the absence of IRS-1, two other pathways, not shared with the IR, originate from the IGF-IR.48 The second pathway originates from Y950 and depends on the activation of the mitogen activate protein kinase (MAPK) pathway, whereas the third pathway originates from serines 1280–3 and requires the mitochondrial translocation of Raf1. Interestingly, Navarro and Baserga51 have found that two of these three pathways are both necessary and sufficient for the mitogenic/survival signal. It does not matter which two pathways are operating, as long as two of them are intact.
Conclusions
The IGF-IR sends contradictory signals to cells.5 It can send a powerful mitogenic and anti-apoptotic signal or it can induce differentiation, which results in growth arrest, differentiation, and cell death. The choice between the two signals seems to depend to a large extent on the availability of substrates. In the absence of IRS-1 (or when concentrations are low), the IGF-IR tends to generate a differentiation signal. When IRS-1 is expressed, the signal is not only mitogenic, but it has the ability to transform cells.32 These contradictions need to be kept in mind if we wish to understand correctly the role of the IGF system in cancer biology.
Acknowledgments
This work is supported by grant CA 78890 from the National Institutes of Health.