Article Text

Abstract
Coeliac disease is the manifestation of an immune hypersensitivity reaction towards gluten and related proteins, in genetically predisposed people. Although the precise pathogenesis of this condition remains to be fully elucidated, it is probably multifactorial in origin. The diagnosis of coeliac disease has traditionally depended on intestinal biopsies alone; nowadays, the diagnosis has been expanded to include an array of serological markers. This review is intended to offer pathologists an update of the relevant history and immunopathology pertaining to coeliac disease and also to offer recommendations on the ongoing responsibilities of the pathologist in the diagnosis and reporting of coeliac disease.
- HLA, human leucocyte antigen
- IELs, intraepithelial lymphocytes
- NK, natural killer
- Th, T helper
Statistics from Altmetric.com
Coeliac disease—also known as coeliac sprue, non-tropical sprue, idiopathic sprue, idiopathic steatorrhoea and gluten-sensitive enteropathy—is a chronic immune-mediated disorder occurring in genetically predisposed people and manifested by a hypersensitivity towards wheat gluten and related derivatives of barley and rye. Afflicted people may develop enteropathy associated with symptoms of bloating, diarrhoea or the sequelae of malabsorption; conversely, many patients remain subclinical. There has been considerable progress in this discipline in recent years, prompting many to reconsider the role of the pathologist in the diagnosis of coeliac disease. Herein, we deal with the ongoing responsibility of the pathologist in the diagnosis and grading of these lesions, in addition to offering a brief summary of the relevant history and immunopathology pertaining to coeliac disease.
BRIEF HISTORY
Although the earliest clinical accounts of coeliac disease are attributed to Aretaeus in the 1st century AD, it was Gee1 who, in 1888, is credited with depicting a modern appreciation of the clinical findings associated with coeliac disease. Of note, Gee was unable to derive an explanation for its pathogenesis, on the gross morphology of the small bowel.
Initially, access to the small bowel mucosa in humans was primarily restricted to autopsy investigations; thus, early attempts to study these tissues were hampered by autolysis. Paulley2 is widely recognised with providing the first histopathological correlation with coeliac disease; nevertheless, he acknowledged the important contributions of his predecessors, including Beneke3 (1910), Justi4 (1913) and Manson-Bahr,5 who, he indicated, each recognised the presence of inflammation and villous atrophy in the small intestine with coeliac disease. Others have offered similar credit to Thin6 (1890). On reviewing several of the works in English it is difficult to substantiate these claims, largely because the application of current diagnostic criteria are lacking.
Pathogenesis
Presently, coeliac disease is widely regarded as an autoimmune disease that arises from an aberrant immune response towards derivatives of gluten, which is present in wheat, barley and rye, in genetically susceptible people.7,8
Unlike the aforementioned cereals, which are members of the Triticeae tribe,9 oats belong to the Aveneae tribe of Graminease10; thus its prolamin—avenin—is considered to be more genetically disparate and perhaps safe among patients with coeliac disease.11 It has also been proposed that a lower percentage of avenin, relative to the total grain protein, may account for this difference in response.10 Regardless, for reasons including potential contamination of oat products with gluten,12 and the possibility that a small percentage of people with coeliac disease harbour a response to avenin,13 those with coeliac disease are advised to exercise caution with these foodstuffs. Other cereals such as rice and millet are considered to be safer, as their proteins bear even less similarity to those of wheat, rye and barley.
Genetic contributors
The concordance rate for coeliac disease is considerably higher among monozygotic twins (75%) compared with those in dizygotic twins (11%)14; among first-degree relatives the rate is estimated at 10–13%.8,14 Coeliac disease is multifactorial and multigenic in origin.15 Human leucocyte antigen (HLA)-DQ2 (DQA1*05/DQB1*02) is associated with most cases of coeliac disease, whereas HLA-DQ8 (DQA1*0301/DQB1*0302) is present in just a minority of patients.16 In addition to associations with HLA, other non-HLA regions of the genome, such as 5q31–33 seem to confer some risk for coeliac disease.15
Immunological contributors
Assuming an accommodating genetic background, the digestion of wheat, rye and barley may expose the bowel to immunoreactive epitopes that subsequently instigate a maladaptive immune response.16 Presumably, some products remain undigested on presentation to the small bowel,17 in turn contributing to the activation of specific populations of T cells in the mucosa.18 Gluten derivatives—including those arising from deamidation complexes with tissue transglutaminase, as well as others in their native state—with an affinity for binding to DQ2 or DQ8 may activate CD4+ T cells.19 Reactive T cell populations, in turn, orchestrate a response targeting multiple endogenous autoantigens, including, paradoxically, the enzyme tissue transglutaminase,20 enterocytes21 and perhaps other targets. The mechanism by which immunoreactive derivatives breach the mucosal epithelium is poorly understood. Possibilities include causing a reactive increase in the permeability of epithelial tight junctions 21; altering uptake and processing by enterocytes,23 perhaps by extensions of the dendritic cells housed in the lamina propria24 or through sampling by Peyer’s patches; alternatively, the response may be instigated by activated intraepithelial lymphocytes (IELs).
A diverse population of immune mediators contribute to coeliac disease, including macrophages, plasma cells, CD4+ T helper (Th) cells, CD8+ cytotoxic T cells and natural killer (NK) cells.25,26 Reports on whether cytokine phenotypes in coeliac disease can be subcategorised based on the Th1/Th2 paradigm are conflicting; however, recent trends would seem to favour a Th1 cytokine bias.27 T lymphocytes in the lamina propria are frequently CD4+. Although there are some CD4+ IELs, most IEL T lymphocytes are CD4−CD8+ and contain the α/β TcR; a minority are CD4−CD8− and express the primitive γ/δ TcR. Some may express NK-type markers, such as CD94 or NKG2D.28,29 The precise role of IELs in coeliac disease is unclear. Cells expressing either the α/β+ or γ/δ+ TcR are up regulated in active disease; on withdrawal of the offending stimulus α/β+ T cells seem to regress, whereas γ/δ+ T cells have a less precipitous denouement.30
Initially it was thought that exogenous gluten products were directly toxic to the mucosa in coeliac disease.9 In contrast with earlier suggestions, IELs are now thought to actively contribute to mucosal damage. Antigen exposure in coeliac disease causes rapid in situ activation of α/β T cell IELs.31 These cells may then damage enterocytes through contributions from several possible mechanisms, including the NKG2D-major histocompatibility complex class I chain-related gene A pathway,29 Fas–Fas ligand pathway,32 perforin-granzyme procedure31 or matrix metalloproteinases.32 Interleukin (IL)15 contributes notably in this response.34,35 The production of IL15 by epithelial cells or macrophages35 may, for example, increase major histocompatibility complex class I chain-related gene A ligand expression on epithelial cells to facilitate the NKG2D receptor signalling pathway.29,36
Plasma cells are present in the lamina propria, but the extent to which the humoral response contributes to the pathogenesis of coeliac disease remains unclear. Multiple types of antibodies have been isolated from patients with coeliac disease. Curiously, a case report detailing coeliac disease in a patient with adult-onset hypogammaglobulinaemia is often cited as evidence that a humoral response is not necessary for coeliac disease.37 Nevertheless, by virtue of the omnipresence of some form of humoral response in coeliac disease, it is not unreasonable to infer a contribution to its pathogenesis, perhaps from the action of activated immune complexes38 or by means of an inhibitory effect on crypt epithelial cell differentiation.39 Fortuitously, antibodies now contribute notably to the diagnosis of coeliac disease, and in monitoring a response to treatment.
DIAGNOSIS
Criteria for the diagnosis of coeliac disease vary. A report from the United European Gastroenterology Week40 emphasised the importance of small bowel biopsy in the diagnosis of coeliac disease, stating “The finding of circulating antibodies. .. supports the diagnosis but is not essential, and should not be used for diagnosis without histologic confirmation.” In people suspected of having coeliac disease, the American Gastroenterological Association mandates a biopsy to confirm the diagnosis.41 Although many still regard the small bowel biopsy as a “gold standard” in the diagnosis of coeliac disease,41,42 the US National Institutes of Health43 recently released a consensus statement recommending biopsies only after a positive serology or when faced with incongruous or indeterminate serological results.
Box 1 Summary of major features of mucosa suggestive of coeliac disease
-
Proximal small bowel involvement, decreasing distally
-
Patchy distribution, in some cases
-
Mucosal architectural changes, including
Villous atrophy
Crypt hyperplasia
Thickening of the basement membrane under the surface epithelium
Reduced numbers of goblet cells
-
Mucosal inflammation
Increased intraepithelial lymphocytes
Influx of immune cells in the lamina propria
-
Enterocyte changes
Cuboidal morphology
Loss of basal nuclear orientation
Cytoplasmic vacuoles
Box 2 Differential diagnosis of small bowel biopsy specimens sharing features of coeliac disease
-
Increased intraepithelial lymphocytes
Allergies to proteins other than gluten (eg, chicken, cow’s milk, eggs, fish, rice and soy; entities cause both raised intraepithelial counts and villous architectural changes)
Autoimmune conditions, various (eg, systemic lupus erythematosus)
Bacterial overgrowth
Blind loop syndrome
Dermatitis herpetiformis
Giardiasis
Graft-versus-host disease
Helicobacter pylori
Inflammatory bowel disease
Irritable bowel syndrome
Microscopic colitis
Non-steroidal anti-inflammatory drugs
Tropical sprue (entities cause both raised intraepithelial counts and villous architectural changes)
Viral enteritis
Crypt hyperplasia or villous flattening
Allergies to proteins other than gluten (eg, chicken, cow’s milk, eggs, fish and soy; entities cause both raised intraepithelial counts and villous architectural changes)
Autoimmune enteropathy
Collagenous sprue
Common variable immunodeficiency
Drug-induced
Hypogammaglobulinaemic sprue
Ischaemia
Kwashiorkor
Radiation therapy
T cell lymphoma, associated enteropathy
Zollinger–Ellison syndrome
Histopathologically, the protean manifestations of coeliac disease (box 1) display a range in severity. An ever-expanding array of entities is known to produce similar histopathological findings (box 2), thereby potentially complicating the histopathological diagnosis of coeliac disease. For this reason, some prudently suggest that the pathologist’s role should remain more indirect, affording the clinician a descriptive report indicating that the lesion is “.. . consistent with CS [celiac sprue]”.44 Depending on the level of serological antibody titres, others advocate abandoning biopsies altogether.45 Presently, we consider the small bowel biopsy essential for diagnosing coeliac disease; likewise, we also acknowledge the multitude of entities that may present with similar histological findings. For this reason it is imperative that there be candid and regular communication between the clinician and the pathologist. Although not always feasible, the clinical–pathological conference can be an ideal venue for such an exchange.
A modest literature proposes a diagnostic role for nasal or rectal biopsies following gluten challenge.46,47 It has been acknowledged that such a technique may afford limited diagnostic value in the context of existing clinical tools, without further investigation.48 For the time being, pathologists are unlikely to encounter such specimens outside an experimental setting.
Normal variations in small bowel histology
The small bowel exhibits a range of morphological variation that should be considered to be normal both among individuals and across populations. A brief overview of normal histological variants follows (for a comprehensive review, see Segal and Petras49). Natural differences in villous architecture across populations can be dramatic, and it is important that pathologists appreciate these differences. Variants of villous morphology include
-
finger-like, with a cylindrical core and rounded apex;
-
leaf-like, with a broad flattened base with a tapering apex;
-
tongue-like, with a broad flattened base and rounded apex; and
-
ridge-like, with a flat linear base that is less in width than its height.
In the duodenum it is not unusual to see branched villi or villi containing fused tips.50 Mixed populations of villi are common.49
Intraindividual differences
In adults, the proximal villi of the duodenum are broad, and a leaf-like morphology is common. In the upper jejunum, tongue-like villi are often seen. More distally, the villi gradually become elongated, assuming a finger-like morphology.51
Although villi overlying Brunner’s glands in the duodenum may exhibit a finger-like appearance, these are typically shorter in length.50 Similar villous changes may be seen in those overlying lymphoid aggregates; in fact, villi may be absent in these areas altogether.50 A larger concentration of IELs may be seen over lymphoid aggregates.
The pathologist should also be beware that gastric metaplasia, gastric heterotopia and heterotopic pancreas can be observed within the small bowel.49
Differences among populations
The developing fetus first develops finger-like villi, followed by crypts.52,53 Exposure of the neonate mucosa to ingested material and intestinal flora results in blunting of the villi; this is relatively transient among infants in temperate regions and more persistent among those in the tropics.54 It may not be until well into childhood that the finger-like morphology replaces the leaf-like variant, assuming the environment is conducive to this change. Yet, the broader villi may represent a normal observation in the proximal small bowel.54 In adults, villous morphology does not seem to change in elderly people.55
The finger-like morphology may predominate in people residing in temperate areas, whereas those indigenous to more tropical climates often have the leaf-like and ridge-like variants proximally and the finger-like variant distally. In its mildest form, this may represent a normal histological variant.56 Interestingly, tropical migrants to temperate regions exhibit some reversal of villous blunting,57 whereas those moving to tropical regions incur villous blunting.58 Although differences in intestinal flora have been proposed as an explanation for loss of the finger-like variant, these differences may also relate in part to dietary differences.56
Differences attributable to sampling
The volume of case material derived from small bowel biopsies is ever increasing, owing in large part to the relative ease of performing upper gastrointestinal endoscopy and procuring tissue. We have found conflicting results on the benefit of suction capsule versus endoscopic pinch biopsies.59,60 Regardless, endoscopic pinch biopsies of the duodenum have largely replaced suction capsule biopsies of the jejunum.
There is evidence supporting a strong correlation in the histological observations in coeliac disease, between the duodenum and jejunum.61 Larger biopsy specimens are generally considered to be more amenable to histological evaluation,41,47,62 although “jumbo” forceps have been reported to confer no marked advantage over a standard bite size.63
Endoscopic biopsies have the advantage of targeting grossly abnormal regions of the bowel to efficiently procure multiple biopsy specimens. Newer endoscopic methods, such as push enteroscopy and double-balloon enteroscopy, allow access to the entire length of small bowel to biopsy forceps. When using pinch biopsy forceps, the endoscopist must be careful not to leave the specimen within the bowel or lose it to the suction chamber. Biopsy forceps crush and destroy tissue and thus evaluation of specimen margins should be limited; in addition to damaging cells, this mode of tissue procurement may introduce artefactual haemorrhage in the sample. Superficial biopsy specimens lacking a muscularis mucosa can cause artefactual separation of the villous bases, resulting in the appearance of shorter and thicker villi.49
Tissue derived from a suction capsule may incur partial or even complete separation of the epithelium from the lamina propria. The absence of a host response to ulceration, such as an acute inflammatory response or granulation tissue, will alert the pathologist to potential sampling difficulties. Due care must be exercised in removing samples from the capsule.50
Histopathology of coeliac disease
Symptomatology in coeliac disease seems to be related to the length of affected bowel, and not to the severity of the mucosal lesion.64,65 Thus, insults compromising the inherent compensatory ability of the small bowel—such as worsening extent of disease, infection, ischaemia and short bowel, among other things—may suffice to unmask previously compensated coeliac disease. Conflicting reports exist on the distribution of lesions in coeliac disease along the small bowel mucosa. It has been suggested that villous lesions rarely coexist with histologically normal mucosa.66 Others describe coeliac disease as exhibiting a patchy distribution,32,50,67,68 thus implying a need for multiple biopsy specimens to secure a diagnosis. Traditionally, the severity of intestinal pathology is regarded as greatest in the proximal small bowel with distal lessening. Lesions affect the mucosa and the submucosa.4 Coeliac disease has also been reported to affect other mucosal sites, such as the oesophagus, stomach and large bowel.
Healing of the small bowel mucosa proceeds in a caudal to cephalad direction.47 This may take anywhere from 6 to 24 months after induction of treatment and in some cases the extent of recovery may remain incomplete.69 The migratory rate of epithelial cells from the crypts to the villous surface is reduced from 3–5 to 1–2 days in coeliac disease.41
Lesions can be described in terms of a range of architectural, cytological and ultrastructural features (table 1) that, combined, create a blurring array of histopathological permutations. Alone, these features are non-specific and should be regarded as shared among a range of ailments afflicting the small bowel (box 2). When screening small bowel biopsy specimens several features may be suggestive of coeliac disease, such as increased IELs, crypt hyperplasia and villous atrophy.67,70
The modified Marsh–Oberhuber classification
Intraepithelial lymphocytes
Because of the nature of the immunopathological basis of coeliac disease, the pathologist encounters a complex and heterogeneous population of lymphocytes and granulocytes in the mucosal biopsy specimens. The lamina propria is the seat of a brisk immunological response, and grading this reaction is difficult and impractical. Fortunately, early work in a murine model showed IELs to be a surrogate marker of immune activity in the lamina propria.71 Researchers in the 1970s extended these results to humans72 and it is from this work that current standards for IEL counts were derived.
Traditionally, counts >40 IELs per 100 epithelial cells were considered to be abnormal.72 This benchmark was subsequently challenged, with 12 IELs per 100 epithelial cells offered as the actual value.73 More recent estimates include 25 IELs,74,75 22 IELs76 or 20 IELs per 100 epithelial cells.77 The trend towards a lower “normal” number of IELs is reflected by the proposal that 30 IELs per 100 epithelial cells were incorporated into revised classification schemes.42 It remains to be determined whether lowering the upper limit for IELs will adversely decrease the specificity of the small bowel biopsy in the diagnosis of coeliac disease; nevertheless, it is conceivable that this number may be further reduced in the future.
Formal enumeration of IELs includes selecting suitably oriented villi in the biopsy specimens and then counting the total IELs present per 100–1000 epithelial cells along the luminal margin, excluding the crypt. The total number of IELs is expressed relative to 100 epithelial cells. For pathologists this procedure is obviously labour intensive, prompting some of them to consider more efficient means of enumerating IELs.75,78
Quantifying villous tip IELs
A rapid method of screening for coeliac disease includes counting the number of IELs present at the villous apex.78 Basically, the pathologist selects five villi. At the distal apex of each villous, 20 epithelial cells are counted and the number of IELs in this range is similarly enumerated; if desired, the number of IELs can then be expressed relative to 100 epithelial cells. This technique has recently been corroborated by others as an efficient means of objectively quantifying IELs,77,79 and the normal number of IELs at the apex per 20 epithelial cells have been documented as 2.2 (11 IELs per 100 epithelial ells),78 4.6 (23 IELS per 100 epithelial cells)75 and 2.3–3.3 (11.5–16.5 IELs per 100 epithelial cells).79 Given the number of entities causing an increase in IELs, an increased number of IELs at the villous tip is by no means diagnostic of coeliac disease, but should raise this possibility in the differential diagnosis.
Loss of the “crescendo sign”
The normal distribution of IELs along the villi assumes a characteristic “luminopetal” distribution,72 that has recently been likened to that of a musical “crescendo”, with a tapering in IELs as we progress towards the villous apex.64 Patients with coeliac disease, including those with architecturally normal villi, lack this pattern as a result of saturation of the tip by lymphocytes, resulting in a more uniform distribution of IELs along the villous length.64,78 A Gestalt approach is applied in assessing biopsy specimens by this method of screening and an abnormal distribution of IELs should alert the pathologist to the possibility of coeliac disease. Despite a rate of potential false positives approaching 25%, this may be more sensitive than formal IEL counts in screening for coeliac disease.78
Immunohistochemistry
Accurately quantifying IELs can be complicated by issues such as nuclear overlap and heterogeneity in nuclear shapes, occasionally making it difficult to distinguish epithelial cells from enterocytes and granulocytes.80 The routine application of immunohistochemical staining for lymphocyte markers such as CD3 has been proposed as a means to better evaluate the number and distribution of IELs in instances in which there is a normal villous architecture and a perceived increase in IELs.77,80
Presently, the benefits of a more rigorous IEL count do not seem to supersede the additional expense, demand on pathologists’ time and delay in issuing of reports to clinicians imposed by immunohistochemistry. When faced with intraepithelial lymphocytosis on haematoxylin and eosin staining, it is perhaps not unreasonable to first seek a second opinion, offer a differential diagnosis or recommend serological testing.
Crypt hyperplasia
Crypt hyperplasia denotes elongation of the length of the crypts of Lieberkühn, a process that initially precedes villous atrophy.65,81 Elongation may be caused by expansion of the lamina propria as a result of the proliferation of stromal cells,81 an influx of inflammatory cells50 and tissue remodelling. The crypts contain stem cells capable of renewing enterocytes and goblet cells, and it is not uncommon to see appreciable mitotic activity in this location. This, unfortunately, is not a reliable indicator of crypt hyperplasia.82 Initial studies investigating the application of proliferative markers such as Ki-67 (MIB-1) in discriminating early stages of coeliac disease have shown some promise.82
The normal ratio of villous height to crypt depth is the subject of some controversy. It is generally assumed that the normal range is 3:1 to 5:149; others have considered other ratios to be acceptable, including 2:1,83 1.82:174 and even 1:1.69 In children, a ratio of 2:1 is considered to be normal by some.84 In coeliac disease, a loss of villous height and elongation of the length of crypts may change this ratio. It remains to be determined whether subcategorisation based on grades of severity is indicated.
Villous atrophy
A loss of villous height is considered to be pathognomonic for coeliac disease by many clinicians; thus, it is important to emphasise the non-specific nature of this finding (box 2). The height of the villous is generally three times its base width. Oberhuber et al67 proposed a grading as mild, marked or total. An absence of atrophy implies that the villi are of normal height. Mild atrophy indicates a minor to moderate amount of villous blunting; marked atrophy indicates the presence of truncated villous remnants; and finally, total atrophy implies the complete absence of villi.67,81 It is important that the most severely diseased areas of the biopsy be reported (ie, biopsy results should not be averaged). Given the potential for an irregular distribution of disease, a comment denoting the patchy nature of the lesion may also be indicated.
The observation of moderate to total villous atrophy, particularly in a patient with longstanding coeliac disease or in a patient who proves unresponsive to diet, obliges the pathologist to attempt to exclude the presence of a more sinister concomitant lesion, such as Crohn’s disease, autoimmune enteropathy, lymphoma or adenocarcinoma.
Additional histological observations
Several additional cytological features can be appreciated in coeliac disease. Enterocytes, for example, may lose their columnar configuration, yielding to a cuboidal shape. The cytoplasm may be basophilic and in some cases contain apical vacuoles.50,67,85 Furthermore, the nuclei may be pyknotic and lose their basal orientation.50 Ultrastructurally, there are reductions in microvillous height with the appearance of apical lysosomes85 and changes in intraepithelial tight junctions.22 Functionally, there are thought to be differences in the glycocalyx or mucous layer of the bowel lumen, resulting from altered patterns of glycosylation; this may selectively promote bacterial adhesion.86
In coeliac disease, the lamina propria may undergo marked expansion, particularly with lymphocytes and plasma cells. Macrophages, eosinophils and mast cells are often seen and occasional neutrophils may also be present. The presence of cryptitis or crypt abscesses should reflexively prompt consideration of the possibility of Crohn’s disease.
APPROACH TO SPECIMEN
Gross specimen
Given the heterogeneous distribution of lesions in coeliac disease and normal differences in small bowel histology, three to five biopsy specimens are recommended. It is important that the clinician provides a description of the location and gross appearance of the area sampled.67 Further, specimens are best oriented by the clinician67; thus, pathologists should offer their colleagues advice in this regard. If necessary, specimens may later be oriented using a dissecting microscope.50
The sample should be handled gently with gloved hands, or smooth or rubber-tipped forceps. Foam pad inserts for specimen cassettes, dental wax or lens paper can be used to orient the specimen before placing it in a fixative. Specimens must be embedded perpendicular to the plane of section50; poorly oriented specimens may contain tangential sectioning, reflected by stratification of either the surface epithelium or crypts. In sectioning from the paraffin-wax block, it is often convenient to cut a ribbon of tissue to minimise unnecessary curling and concomitant manipulation of the specimen.
Fixation artefact, most notoriously derived from (a) exposing the specimen to air for a prolonged period or (b) freezing the tissue for quick sectioning, can markedly distort tissue histology. For this reason, tissue should be kept moist and placed promptly in a fixative. The freezing of routine small bowel biopsy specimens is discouraged, unless indicated for purposes such as immunohistochemical analysis. This may be reduced by adding glycerol to the fixative. Some fixatives may also introduce histological changes; to limit difficulties in interpretation, we use a standard 10% neutral-buffered formalin solution. Some pathologists may prefer other fixatives—such as Bouin’s, Carnoy’s or B-5—that yield equal or superior histology; however, many can be difficult to use or may contain heavy metals. Differences in interpretation may also arise from poor standards in histochemical staining. It is important that the pathologist be acquainted with the techniques used in their laboratories and the means by which they can introduce subtle improvements in their laboratory’s staining protocols, if necessary.
Microscopic specimen
The pathologist will routinely assess the following:
-
villous architecture, including the crypt to villous ratio;
-
crypts;
-
lamina propria;
-
muscularis mucosae;
-
enterocytes;
-
the composition of the inflammatory cell infiltrate in the epithelium and lamina propria, particularly intraepithelial lymphocytosis;
-
the brush border; and
-
the lumen border (eg, for evidence of infection).
To assess for villous atrophy, the specimen must be properly oriented to disclose a series of at least four complete villi.87 If these are absent, it is prudent to make a comment in the pathology report. If indicated, the pathologist may also consider requesting that the specimen be re-embedded.
Histopathological classification
One of the first attempts to histopathologically classify coeliac disease was proposed by Rubin et al.50 Considering features such as epithelial surface area, as reflected by villous height, as well as epithelial abnormalities and infiltration of the lamina propria by inflammatory cells, they proposed that lesions be assessed as mild, moderate or severe.50
Arguably, the most important advance in classifying lesions in coeliac disease was forwarded by Marsh,70,88 who proposed a series of interrelated lesions that integrates the pathophysiology of coeliac disease with its histopathological correlates. The Marsh grading system outlines four categories of lesions associated with coeliac disease: pre-infiltrative (type 0), infiltrative (type 1), infiltrative-hyperplastic (type 2) and flat-destructive (type 3) 70; the atrophic-hypoplastic (type 4) lesion appears in later publications.88,65 This system of grading assesses the presence of an immune response in the epithelium and describes the degree of architectural changes in the mucosa. This classification was revised to facilitate diagnostic applications,42,67,89 as the Marsh–Oberhuber grading system,90 which subcategorised type 3 lesions based on villous height as type 3a mild atrophy, type 3b marked atrophy and type 3c total villous atrophy.89 This classification is widely used by pathologists today.90
Marsh–Oberhuber: type 0 lesion
These specimens contain histologically normal small bowel mucosa.67 The villous architecture is unadulterated by villous blunting or crypt hyperplasia; similarly, there are fewer than 30 IELs per 100 epithelial cells. Patients in this group may be identified based only on serological criteria and may remain clinically silent (table 1).
Marsh–Oberhuber: type 1 lesion
These lesions are characterised by architecturally normal small bowel mucosa and contain intraepithelial lymphocytosis with >30 IELs per 100 epithelial cells.42,61 An ever increasing number of entities are recognised as causing intraepithelial lymphocytosis (box 2), making this observation relatively non-specific. In the absence of a clinical or family history or serological evidence of coeliac disease, this observation is suggestive, but not diagnostic, of coeliac disease (table 1; fig 1). Increased IELs with an otherwise unremarkable villous architecture may occur in as many as 10% initially presenting with coeliac disease.91

Type 1. Normal villous architecture with increased intraepithelial lymphocytes. Haematoxylin and eosin, ×100.
Sample pathology report
Diagnosis: Intraepithelial lymphocytosis with normal villous and crypt architecture. Comment: These histopathological features are non-specific and clinical correlation is indicated.
Marsh–Oberhuber: type 2 lesion
Architecturally, these intermediary lesions maintain a normal villous architecture but contain crypt hyperplasia81; this is coupled with an intraepithelial lymphocytosis of >30 IELs per 100 epithelial cells.52,61 Rarely observed in a clinical setting,67 the utility of this category has recently been challenged.90 Indeed, there is disagreement in the literature on defining crypt hyperplasia, difficulty in quantitating this phenomenon67 and little information to support a clinical correlate; until resolved, we advocate continuing to consider the range of 3:1 to 5:1 as normal. As with type 1 lesions, the presence of a type 2 lesion alone is sufficiently non-specific to not immediately elicit the diagnosis of coeliac disease. Clinically, it may, for example, be seen in patients with coeliac disease who are undergoing treatment, or in dermatitis herpetiformis (table 1).67
Sample pathology report
Diagnosis: Intraepithelial lymphocytosis with crypt hyperplasia and normal villous height. Comment: In addition to intraepithelial lymphocytosis, there is crypt hyperplasia. The villi exhibit a normal finger-like appearance, with no marked blunting. These histopathological features are non-specific and clinical correlation is indicated.
Marsh–Oberhuber: type 3 lesion
Type 3 lesions are characterised by increased numbers of IELs (>30 IELs per 100 epithelial cells), crypt hyperplasia and villous atrophy.42,61,67,81 Marsh’s65,70 original classification does not differentiate the extent of villous blunting. Subclassification of Marsh’s type 3 “flat-destructive lesion” was proposed to differentiate differences in the degree of villous atrophy as partial (3a), subtotal (3b) and total (3a) villous atrophy.89 To limit confusion, Oberhuber et al67 in turn proposed mild, marked and total villous atrophy (flat mucosa) to describe the respective subcategories. The term “moderate” in lieu of “marked” villous atrophy is not unreasonable, and may prove to be more convenient and consistent with recent trends in classification (table 1).
Thus, in addition to increased numbers of IELs (>30 IELs per 100 epithelial cells) and crypt hyperplasia, type 3 lesions can be subcategorised on the basis of villous atrophy into type 3a (mild), with minor villous blunting (fig 2); type 3b (moderate), with intermediate villous atrophy (fig 3); and type 3c (total), with a flat mucosa and no visible villi (fig 4). Poorly oriented specimens may result in failure to appreciate subtle differences in villous height8; this may be particularly problematic in distinguishing type 3a and 3b lesions.
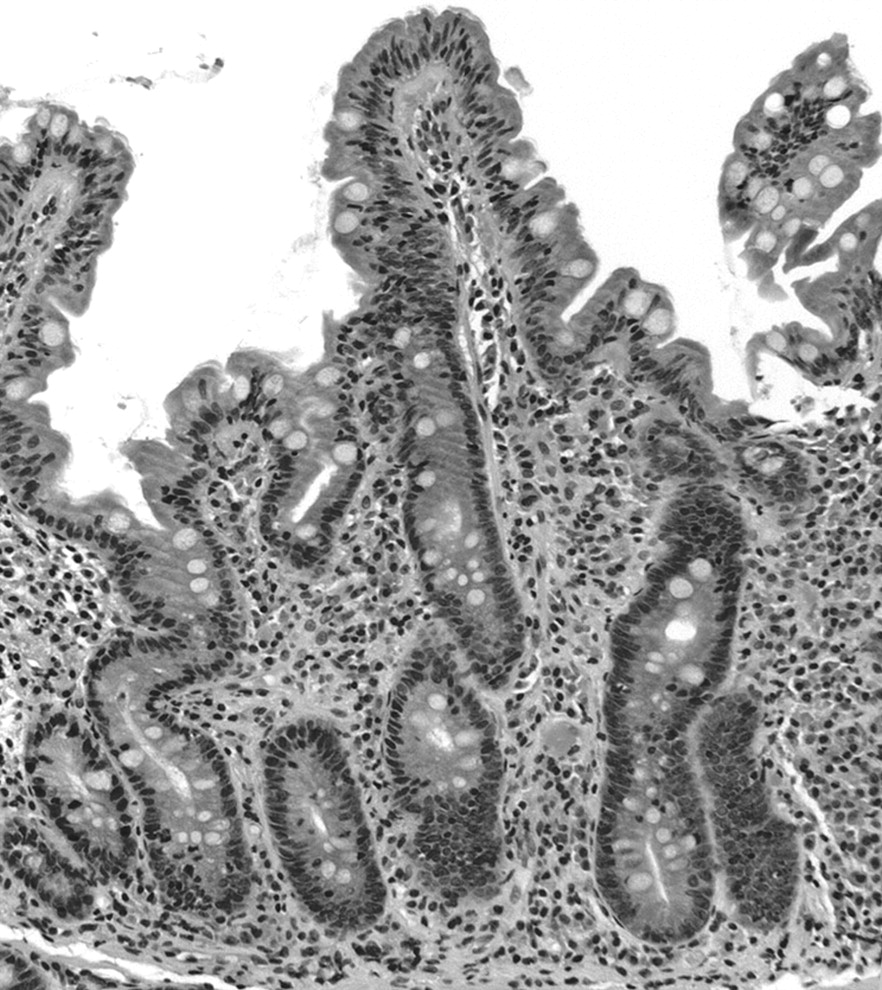
Type 3a. Increased numbers of intraepithelial lymphocytes, crypt hyperplasia and mild villous atrophy. Haematoxylin and eosin, ×100.

Type 3b. Increased numbers of intraepithelial lymphocytes, crypt hyperplasia and moderate villous atrophy. Haematoxylin and eosin, ×100.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Type 3c. Increased numbers of intraepithelial lymphocytes, crypt hyperplasia and total villous atrophy. Haematoxylin and eosin, ×100.
Sample pathology report
Diagnosis: Intraepithelial lymphocytosis with crypt hyperplasia and moderate villous atrophy, suggestive of coeliac disease. Comment: In addition to intraepithelial lymphocytosis, there is crypt hyperplasia. The villi exhibit a leaf-like appearance, with moderate blunting in height. Although these histopathological features are not specific, they are suggestive of coeliac disease corresponding to a Marsh–Oberhuber type 3b lesion.
Marsh–Oberhuber: type 4 lesion
These rare lesions lack villi, but contain a normal crypt height and number of IELs.65,67 They are reported to be irreversible and are thought to result from malnutrition67 or as a consequence of aberrant IEL homeostasis.91 It has been proposed that such lesions be removed from this classification.90 The pathologist should be vigilant of lesions that prove unresponsive to withdrawal of offending stimulants, particularly in the light of the recent literature confirming coeliac disease as contributing to multiple intestinal neoplasms, including enteropathy-associated T cell lymphoma, non-Hodgkin’s lymphoma and adenocarcinoma of the small bowel.25,92 Such lesions may mandate more rigorous immunohistochemical analysis90 or consultation with a specialist in gastrointestinal pathology (table 1).
Pathology report
As exemplified above, the diagnosis should include a description of the villous (eg, mild, moderate and total atrophy) and crypt architecture, presence or absence of intraepithelial lymphocytosis and any additional pathological observations. In instances in which the pathologist is confident of rendering the diagnosis suggestive of coeliac disease, it is recommended that the Marsh–Oberhuber grade be included. It is also reasonable to offer a differential diagnosis (box 2); recommendations for serological screening or re-biopsy; or to consult a pathologist with expertise in the field.
Future considerations
It has been suggested that the Marsh–Oberhuber grading system can be further simplified to limit interobserver variation and promote diagnostic reproducibility.90 With this proposal—parenthetically, it bears resemblance to the type 1/type 2 lesions under “Grade A”; combine type 3a and 3b lesions as “Grade B1”; convert type 3c into “Grade B2”; and discard type 4 lesions.90 Although there are no obvious problems with this proposal—it bears resemblance to the early type 1 or type 2 classification initially forwarded by Marsh93— there is a risk that it may prove unnecessarily confusing for pathologists and clinicians, thereby defeating its stated intent. Perhaps at this juncture it is not unreasonable to call for a consensus among gastrointestinal pathologists to establish a standardised reporting approach to coeliac disease.
CONCLUSION
The small bowel biopsy remains an essential component to the screening and diagnosis of coeliac disease. However, a diagnosis on the basis of histopathology alone is complicated by a large number of potential mimics. Similarly, non-invasive means of diagnosing coeliac disease are currently unable to replace the biopsy as a gold standard. In the interest of the patient, the diagnosis of coeliac disease is most soundly achieved through candid communication between the clinician and the pathologist. It necessitates incorporating details of the patient’s history and adjunct investigations, with the histopathological assessment of the small bowel mucosa. The pathology report should offer a brief, albeit descriptive, summary of the salient histological findings in a manner that can readily be assimilated by the clinician. If indicated, the pathologist may recommend serological testing, follow-up re-biopsy or seek the opinion of a colleague with a specialised interest in gastrointestinal pathology. These recommendations are intended to promote standardisation and consensus among pathologists, clinicians and researchers.
Acknowledgments
We thank Dr CT-S Chung and Dr MN Marsh for their thoughtful suggestions on reviewing an early draft of the manuscript.
REFERENCES
Footnotes
-
Competing interests: None.