Article Text

Abstract
Background: Deletions in the β-globin cluster causing thalassaemia and hereditary persistence of fetal haemoglobin (HPFH) are uncommon and difficult to detect. Data in Chinese are very scarce.
Aims: To use a recently available technique to investigate the frequencies and nature of β-globin cluster deletions in Chinese.
Methods: 106 subjects with phenotypes of thalassaemia or HPFH and suspected to have deletions in the β-globin cluster were studied. A commercially available kit employing multiplex ligation-dependent probe amplification (MLPA) was used to screen for deletions. Gap PCR and direct nucleotide sequencing were used to characterise deletions detected.
Results: 17 deletions in the β-globin cluster were found in 17 patients: 8 of Chinese (Aγδβ)0 thalassaemia, 7 of Southeast Asian (Vietnamese) deletion and 2 of Thai (Aγδβ)0 thalassaemia. The only type of deletion detected in δβ-thalassaemia was Chinese (Aγδβ)0 thalassaemia. The deletional form of HPFH was rarely seen in only 1 case of Thai (Aγδβ)0 thalassaemia. Deletions presenting as β-thalassaemia trait and raised HbF were all of the Southeast Asian (Vietnamese) deletion type. When these deletions were co-inherited with classical β-thalassaemia mutations in compound heterozygous states, the phenotypes could be very variable.
Conclusions: In the Chinese population, there are only relatively few types of deletions seen in the β-globin cluster. MLPA is a fast and effective way of screening for these deletions. Characterisation of these deletions allows the development of simpler and more specific PCR-based tests for routine diagnostic use. Accurate prediction of phenotype is not always feasible. The molecular defects in many cases of HPFH still await discovery.
Statistics from Altmetric.com
Defects in the β-globin gene cluster at chromosome 11p15.4 cause thalassaemias, haemoglobinopathies and fetal haemoglobin (HPFH). Deletions greater than 1 kb are seen in deletional HPFH and most δβ-thalassaemias. Deletions are distinctly uncommon in β-thalassaemias, which are usually due to single nucleotide substitutions or insertion/deletion of a few nucleotides in the β-globin gene.1 Screening for larger deletions is technically difficult. Routine PCR-based techniques target only known deletions, while direct nucleotide sequencing of promotors, exons and exon-intron junctions is not helpful in this setting as a normal allele is present. Deletions can be screened by Southern blotting, array comparative genomic hybridisation (CGH), quantitative PCR, fluorescence in-situ hybridisation with tiling probes and conventional cytogenetics. However, these techniques all have disadvantages of low resolution, high cost, poor throughput, or complex test design and data readout. Because of these limitations, the prevalence and nature of β-globin gene cluster deletions in many populations remains uncharacterised. This hinders the development of a comprehensive diagnostic algorithm and the provision of genetic counselling and prenatal diagnosis.
Multiplex ligation-dependent probe amplification (MLPA) is a recently described method that can detect mid-size deletions down to a few hundred bases or single exon level.2 Each probe consists of two oligonucleotides that bind adjacent to each other at the target sequence. A ligation reaction takes place and all intact probes are amplified in a PCR using one single set of labelled primers as all probes have the same primer recognition ends. Amplification products differ in size, which can be separated by electrophoresis using a standard sequencer. The peak height/area of a probe represents the amount of amplification product, which is in turn proportional to the copy number of target sequence recognised by the probe in the sample. This technique is fast and reliable and it can be applied to many hereditary and acquired diseases caused by gene dosage changes.3 We used a commercial MLPA kit to screen a group of Chinese patients suspected to have deletions in the β-globin gene cluster. Deletions detected were further characterised and correlated with phenotypic data. The study results provide information on the prevalence of these deletions in the Chinese population and thus facilitate the development of simple and specific testing protocols.
Methods
Subjects, phenotyping and globin genotyping
A total of 106 Chinese subjects suspected to have deletions in the β-globin gene cluster were identified in our records from 1998 to 2008; 102 were archive clinical samples sent for globin phenotyping and genotyping, and 4 patients were recruited from an ongoing genetic association project. Clinical and haematological data, including blood counts and haemoglobin (Hb) study results, were retrieved. HbA2 and HbF levels were assayed using cation exchange high performance liquid chromatography (1998 to June 2003, Variant; July 2003–06, Variant II, BioRad, Hercules, California, USA). DNA was extracted from peripheral blood samples collected in EDTA using standard phenol/chloroform method. Routine α-globin genotyping (screening for −−SEA, −α3.7 and −α4.2), β-globin genotyping (screening for deletion of CTTT in codons 41/42, IVSII-nt 654 (C→T), nt -28 (A→G), codon 17 (A→T), codon 43 (G→T) and codons 71/72 (+A)), detection of anti-3.7 and anti-4.2 α globin gene triplication, and direct nucleotide sequencing of α- and β-globin gene were performed as previously described at diagnosis in all 106 subjects.4 5 A gap PCR for was set up in our laboratory to detect Chinese (Aγδβ)0 thalassaemia since 2003 and those cases were not included. No routine molecular test for HPFH was established before this study.
There were 46 men and 60 women, aged 4 months to 82 years. They were categorised into seven groups according to phenotype and routine globin genotyping results (table 1).
Classification of 106 patients by phenotype and routine globin genotyping results
In group IV, subjects had raised HbA2 suggestive of β-thalassaemia trait but their HbF with significantly raised (>5%). No β-globin gene mutation was detectable in this group. In group V, subjects harboured a common heterozygous β-globin gene mutation. A concomitant HPFH trait was suspected because of raised HbF. In group VI, patients had a β-thalassaemia intermedia or major phenotype, but a β-globin gene mutation was only found in one of the parents, which was inherited by the patients. None of the patients had a triplicated α-globin gene configuration that could have modified their clinical phenotype. We also included in this study a group of patients with β-thalassaemia intermedia or major phenotype (group VII). Only one common β-globin gene mutation was detected. As no family study was performed, homozygosity was presumed but not confirmed. A deletion of the other β-allele could not be excluded.
Twenty-six archive normal DNA samples with normal red cell indices and haemoglobin study results, and one archive sample with known Chinese (Aγδβ)0 thalassaemia deletion were used as negative and positive controls respectively for validation of the MLPA kit.
Multiplex ligation-dependent probe amplification
Probes and reaction mixture for ligation and PCR were purchased from MRC-Holland (SALSA MLPA kit P102 HBB, MRC-Holland, Amsterdam, the Netherlands). Twenty-five probes spanning a 68.7 kb region of the β-globin gene cluster from 5′ of the locus control region to 3′ of the β-globin gene were used. HS1–4 of the β-locus control region and all coding genes (HBE1, HBG2, HBG1, HBD, HBB) were targeted. Four DNA quantity control probes were included to safeguard adequacy of test DNA, and another ligation-dependent probe targeted at chromosome 2q14 was used to control for successful ligation reaction. Fourteen other control probes targeting different chromosomal sites other than 11p15.4 were present in the probemix for result normalisation in each sample. The SALSA MLPA P102 HBB probemix was designed by Helena Duarte and Jan Schouten at MRC-Holland. A 200 ng aliquot of sample DNA was used for each subject. Tests were performed according to the manufacturer’s instructions using a standard thermocycler (GeneAmp PCR System 9700, Applied Biosystems, Foster City, California, USA). Amplification products were separated by electrophoresis in an ABI-3130XL Genetic Analyzer (Applied Biosystems) and results analysed by GeneMapper v3.7 (Applied Biosystems). A normal control sample was run in every batch of patient samples.
MLPA data analysis
Data from GeneMapper were exported to a web-based program for analysis (Coffalyser MLPA DAT v5.4, MRC-Holland). Global normalisation was used, where all peak areas were normalised by dividing each peak area by the combined peak area of all peaks in a particular sample. From the 26 normal samples, the mean and SD of the normalised peak area for each probe was calculated. Deletion of a probe recognition sequence in a patient sample was defined as a normalised peak area below 3 SD of the mean normalised peak area for that probe in the normal controls. Heterozygous deletion (copy number of one) resulted in a 35–50% reduction of normalised peak area of a probe. Homozygous deletion (zero copy) was seen as complete absence of a peak.
Characterisation of deletions detected by MLPA
The extent of deletions detected by MLPA was compared to known β-globin gene cluster deletions reported in Southeast Asians. Gap PCR was performed to screen for candidate deletions using the respective flanking primers (table 2). Details of reaction conditions are available as supplementary data on the website.
Primer sequences and references
Results
Performance of MLPA
No loss of probe amplification signal (no deletion) was detected in any of the 26 normal control samples. Deletion with an extent consistent with Chinese (Aγδβ)0 thalassaemia was detected in the positive control sample previously confirmed to have this deletion. The DNA quantity controls and ligation-dependent control confirmed sufficient DNA and successful ligation and amplification in all test batches. In five patient samples, reduced peak areas were seen in several isolated single probes. Probes involved were non-random (exon 1 of δ-globin gene and exons 2 and 3 of β-globin gene). Polymorphism or genuine deletion of the probe binding sites was initially suspected. Direct DNA sequencing of these sites using primers derived from the recognition sequences of flanking MLPA probes showed no evidence of polymorphism or deletion in any of the five patients (data not shown). Testing was repeated using a new version of the MLPA kit (SALSA MLPA kit P102-B1 HBB, MRC-Holland) in which the three implicated probes were redesigned. No loss of probe signal was observed, suggesting that the inconsistent amplification of the three probes in the original kit was likely due to suboptimal probe design.
Deletion detected by MLPA
A total of 17 heterozygous deletions were detected in 17 patients, accounting for 16% of 106 subjects studied. All deletions were characterised by gap PCR using primers for known β-globin gene cluster deletions reported in Southeast Asians. Figure 1 shows the extent of the three types of deletion found, in relation to the main regulatory elements at the β-globin gene cluster. Table 3 shows clinical phenotype, haematological data and the final β-globin cluster genotype of these 17 patients.

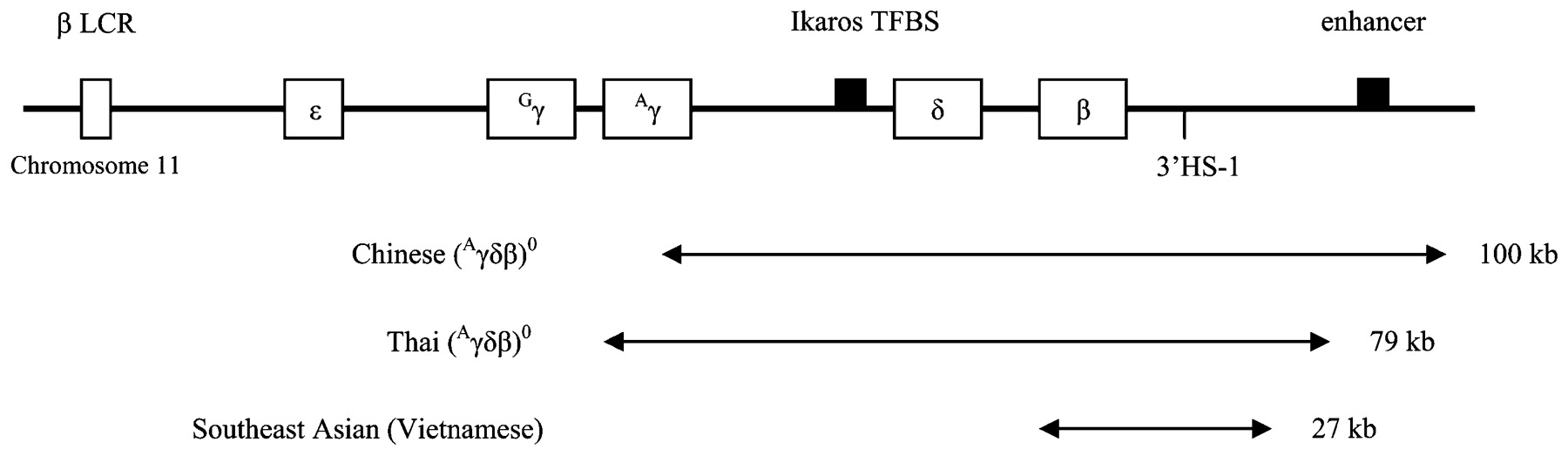{kind=link}
Schematic diagram of extent of the three deletions identified.8 9 10 LCR, locus control region; Ikaros TFBS, Ikaros transcription factor binding site implicated in γβ-globin gene switching; 3′HS-1, 3′ DNAase hypersensitivity region-1 γ-silencing element; enhancer, 3′ erythroid-specific enhancer element.
Clinical phenotype, haematological data and final globin genotype of 17 patients with deletions detected by multiplex ligation-dependent probe amplification
The commonest type was Chinese (Aγδβ)0 thalassaemia9 (n = 8), followed by Southeast Asian (Vietnamese) deletion10 (n = 7) and Thai (Aγδβ)0 thalassaemia (HPFH-6)8 (n = 2). Deletions were mostly found in group II (δβ-thalassaemia trait, 4/8), group IV (β-thalassaemia trait with raised HbF, 5/9) and group VI (β-thalassaemia intermedia/major with β-globin gene mutation detected only in one parent, 5/28). Together they accounted for 14 of 17 deletions detected. Deletions were distinctly uncommon in group I (β-thalassaemia trait, 0/4), group III (HPFH trait, 1/10) and group V (heterozygous β-globin gene mutation with raised HbF, 0/16) (table 2).
In the heterozygous state (groups I to V), the types of deletion detected within each group were homogeneous. All four deletions found in group II (δβ-thalassaemia trait) were Chinese (Aγδβ)0 thalassaemia. All five deletions found in group IV (β-thalassaemia trait with raised HbF) were Southeast Asian (Vietnamese) deletion. The single case of deletional HPFH found in group III was Thai (Aγδβ)0 thalassaemia (HPFH-6). On the other hand, different types of deletions were seen in patients with β-thalassaemia intermedia/major in groups VI and VII, where the deletions occurred in the compound heterozygous state with a co-existing β+ or β0 mutation.
Of note, 2 of 31 β-thalassaemia intermedia/major patients in group VII, who were previously diagnosed to have homozygous β-globin gene mutation using routine globin genotyping methods, were found to harbour a β-globin gene cluster deletion.
Discussion
This is the largest study reporting to date on globin gene deletion in thalassaemia patients using MLPA as a screening tool. Excluding group VII (patients presumably labelled as homozygous for common β-thalassaemia mutations without family study) where the pre-test probability for deletion is the lowest, the deletion detection rate in our cohort is only 20% (15/75). A further 9 patients with δβ-thalassaemia of the Chinese (Aγδβ)0 type were detected in our laboratory by gap PCR since 2003 during the study period. Inclusion of these 9 cases in our cohort would only have increased the prevalence to 28.5% (24/84). This is still substantially lower than the 61% reported in a previous Dutch study using the same method,11 although sample selection criteria are different. These findings suggest that there may be major differences in the prevalence of β-globin cluster deletion among different ethnic groups.
Within our Chinese cohort, relatively few types of deletion are present in defined phenotypic groups. Chinese (Aγδβ)0 thalassaemia is the only deletion detected in δβ-thalassaemia. In the heterozygous state, Southeast Asian (Vietnamese) deletion characteristically gives rise to an otherwise typical β-thalassaemia trait phenotype with microcytosis and increased HbA2, albeit with a significantly raised HbF level as well. This unique phenotype is predictable from its extent of deletion, which includes the β-globin gene and the 3′HS-1 γ-silencing element, but leaves the δ-globin gene and the further 3′ erythroid-specific enhancer element intact (fig 1). If the presence of a common β-thalassaemia mutation has been excluded, Southeast Asian (Vietnamese) deletion should be considered when patients present with this particular phenotype. Only one form of deletional HPFH is seen—Thai (Aγδβ)0 thalassaemia (HPFH-6), which spares the 3′ erythroid-specific enhancer element (fig 1). Knowledge of this homogeneity of β-globin cluster deletions in Chinese and their characteristic phenotypes will facilitate the set-up of a fast, efficient and comprehensive PCR-based protocol for molecular diagnosis of β-globin cluster disorders.
The two patients who are compound heterozygous for Southeast Asian (Vietnamese) deletion and β0-thalassaemia mutation present as β-thalassaemia intermedia (table 3). This argues against it being a deletional HPFH defect as previously suggested,12 but is more akin to a β-thalassaemia phenotype with raised HbF as is seen in all five heterozygous subjects in the present study. However, the phenotypes of compound heterozygotes of Chinese (Aγδβ)0 or Thai (Aγδβ)0 defect with common β0 thalassaemia mutation are less predictable. One of the two patients with identical βIVS2 654(C>T)/βChinese (Aγδβ)0 genotype presents as β-thalassaemia major while the other shows only intermedia severity. The patient who is compound heterozygous for β43(GAG>TAG)/βThai (Aγδβ)0 has thalassaemia major which is much more severe than one will expect from a β0/βHPFH genotype. It is certain that other unidentified modifiers of disease phenotype, such as separate HPFH traits or rarer forms of α-gene duplication, exist to account for the observed phenotypic heterogeneity. Further studies on these complex interactions will not only broaden our knowledge of thalassaemia pathogenesis but can greatly facilitate genetic counselling.
Deletions are not detectable in many thalassaemic subjects in our patient cohort. The underlying molecular defects leading to thalassaemia remain elusive. Non-deletional mutations in the regulatory regions in β-globin cluster can be targets for further investigation, such as the β-locus control region, the Ikaros transcription factor binding site immediately upstream of δ-globin gene which is implicated in γβ-globin gene switching,13 the 3′HS-1 regulatory sequence for γ-silencing14 and the further 3′ erythroid-specific enhancer element.15 We have excluded common triplicated α-globin gene configurations by multiplex PCR, although rarer forms of α-globin gene duplication causing an intermedia phenotype in some β-thalassaemia heterozygotes are still possible.16 The very low detection rate in patients suspected to harbour deletional HPFH is unexpected, as many of them have significantly raised HbF above 10% to even 50%. Further studies to look for non-deletional types of HPFH mutations within the β-globin cluster such as γ-promoter mutations are indicated. Other quantitative trait loci for HbF expression, including chromosome 6q23 and 2p15 region have also been shown in various populations, including Chinese.17 18
Our study clearly shows a role for MLPA in deletion screening in the β-globin cluster. The technique is fast and easy to perform, requiring only standard instruments in a routine molecular diagnostic laboratory. Results analysis is simple. It is relatively inexpensive when compared to other techniques such as Southern blotting, array CGH or quantitative PCR. However, the potential pitfalls must be borne in mind. Theoretically, small deletions of unknown regulatory regions in the β-globin cluster not targeted by the MLPA probes can be missed, as consecutive probes are not continuous or overlapping. Loss of isolated single probes must therefore be interpreted with caution. One has to exclude polymorphism at binding sites of probes and use alternative methods to confirm any suspected small deletions.
In conclusion, using MLPA as a screening tool, relatively few types of deletions are seen in the β-globin gene cluster in Chinese. Characterisation of these deletions allows the development of simpler and more specific PCR-based tests for their detection, a strategy that has been successfully employed for detection of common α-globin gene deletions using a multiplex gap–PCR approach. However, accurate genotype–phenotype correlation is not always feasible. Moreover, the molecular defects in many cases of HPFH still await discovery. Whether these observations can be generalised depends on results of similar large scale studies in other Chinese populations.
Take-home messages
Large deletions in the β-globin gene cluster are relatively uncommon in Chinese, particularly in the group of hereditary persistence of fetal haemoglobin.
A narrow spectrum of deletions is observed, with each deletion showing a characteristic phenotype.
However, an accurate genotype–phenotype prediction is not always possible in the compound heterozygous states.
Multiplex ligation-dependent probe amplification is a good screening method for unknown β-globin gene cluster deletions.
Acknowledgments
We thank Professor David H K Chui for permission to include in this study four subjects from an NIH/NIDDK funded project.
REFERENCES
Supplementary materials
web only data 62/12/1107
Files in this Data Supplement:
Footnotes
Supplementary data is published online only at http://jcp.bmj.com/content/vol62/issue12
Funding This work was supported by a grant from the Children’s Thalassaemia Foundation of Hong Kong (project no. 2007/03).
Competing interests None.
Ethics approval Not applicable. Reason: All 102 patient samples were initially taken with proper consent for globin study during diagnostic workup which includes phenotyping and genotyping. The four subjects recruited from an ongoing genetic association study have given written consent and ethical approval has been obtained for genotyping.
Provenance and peer review Not commissioned; externally peer reviewed.