Article Text

Abstract
Aim—To describe six patients with pulmonary marginal zone lymphoma in whom amyloid deposition was identified. Marginal zone lymphoma is a recently recognised type of low grade non-Hodgkin's lymphoma.
Methods—A computerised search was performed of all patients seen at the Mayo Clinic with a diagnosis of pulmonary amyloidosis. Six patients with pulmonary amyloidosis who had biopsy confirmed extranodal marginal zone lymphoma of mucosa associated lymphoid tissue type were identified. All were women, ranging in age from 45 to 85 years.
Results—Five patients had amyloid deposition in conjunction with pulmonary marginal zone lymphoma at the time of the original diagnosis. One patient was referred for evaluation of localised pulmonary amyloidosis and was found to have coexisting pulmonary marginal zone lymphoma. Clinical presentation was limited to pulmonary symptoms (two of the six) or constitutional symptoms (two), or was asymptomatic (two). In all six cases, initial findings of nodular densities on screening chest roentgenograms led to further evaluation and eventual lobectomy; these findings included multiple pulmonary nodules (four), single nodule (one), and single nodule with diffuse bilateral interstitial infiltrates (one). Bone marrow was examined in five patients and was normal in all. Protein studies performed in four patients revealed no monoclonal protein. No patients had manifestations of systemic amyloidosis, such as renal, neurological, or cardiac involvement, at a median follow up of 50 months. Four of the six patients remain alive at a median of five years.
Conclusions—Pulmonary marginal zone lymphoma may be found in association with localised amyloid deposition and should be considered in the differential diagnosis of localised pulmonary amyloidosis.
- marginal zone lymphoma
- non-Hodgkin's lymphoma
- amyloidosis
Statistics from Altmetric.com
Pulmonary amyloidosis is a disorder characterised by the deposition of insoluble monoclonal immunoglobulin light chain fragments in lung tissue. The deposition of amyloid in the respiratory tract is common in systemic amyloidosis, either in primary systemic disease or secondary to systemic disease processes such as chronic renal failure, chronic infections, rheumatoid arthritis, tuberculosis, syphilis, osteomyelitis, or inflammatory bowel disease.1 Localised amyloid deposition in lung tissue is a well described but less frequently observed form occurring in characteristic tracheobronchial, nodular parenchymal, or diffuse interstitial patterns. Findings are not associated with systemic amyloid involvement.1–9 In three papers,10–12 localised deposition of amyloid was found in association with gastrointestinal marginal zone lymphoma but no similar association with pulmonary marginal zone lymphoma has been reported. Herein, we describe six patients with localised pulmonary amyloidosis in whom biopsy confirmed pulmonary marginal zone lymphoma was found at the time of the original diagnosis.
Methods
A computerised search was performed of all medical records of patients seen at the Mayo Clinic with the diagnosis of pulmonary amyloidosis. We then identified patients in whom the diagnosis of coexisting pulmonary marginal zone lymphoma of mucosa associated lymphoid tissue (MALT) type had been made on the initial lung biopsy specimen, or after a primary diagnosis of pulmonary amyloidosis. The patient records and histopathological material were reviewed for diagnoses of pulmonary amyloid and marginal zone lymphoma.
The diagnosis of amyloid deposition was made by Congo red staining of tissue sections and the demonstration of congophilic material that showed apple green birefringence under polarised light. In addition, sufficient tissue was available to perform immunohistochemistry for amyloid P component and free immunoglobulin light chains. In all three cases, light chain amyloid was demonstrated. The diagnosis of extranodal marginal zone lymphoma of MALT type was made by using the criteria of the revised European–American lymphoma (REAL) classification.13 Only patients with marginal zone lymphoma and amyloid deposition on initial lung biopsy specimens were included. Immunoperoxidase stains were performed on three lung samples in which adequate tissue remained in the paraffin wax block, using the method of Kurtin et al.14 In brief, the slides were dewaxed and hydrated. Antigen retrieval was performed by using protease (Ventana Medical Systems, Tucson, Arizona, USA) for the κ and λ immunoglobulin light chain stains (polyclonal antibodies; Dako Corporation, Carpinteria, California, USA), 44% formic acid for amyloid P component (polyclonal antibody; Calbiochem, San Diego, California, USA) steaming in 1 mM citrate buffer, pH 6.0, for CD20 staining (L26; Dako), or steaming in EDTA buffer, pH 8.0, for CD3 (polyclonal antibodies; Dako). The slides were then stained for κ and λ light chains, CD20, and CD3 with the use of a Ventana automated instrument and the labelled streptavidin biotin peroxidase technique, according to the manufacturer's protocols.
Clinical and laboratory data were abstracted from the patients' records. Particular attention was focused on clinical findings suggestive of extrapulmonary or systemic amyloid disease, including renal, cardiac, neurological, hepatic, dermatological, and musculoskeletal evaluations. Renal amyloidosis was defined by the minimum criteria of nephrotic range proteinuria with or without associated hypoalbuminaemic oedema. Amyloid cardiomyopathy was defined by the minimum criteria of diastolic dysfunction (early disease), overt congestive heart failure, or amyloid detection on two dimensional echocardiography. Amyloid neuropathy was defined by the minimum criteria of upper or lower extremity paresthesias without other known aetiology (early disease), autonomic neuropathy (orthostatic hypotension, autonomic diarrhoea, loss of bladder control, or impotence), carpal tunnel syndrome, or demonstrable amyloid deposition on sural nerve biopsy. Hepatic amyloidosis was defined by the minimum criteria of hepatomegaly, raised liver enzymes, and monoclonal protein in serum or urine. Other criteria for extrapulmonary or systemic amyloidosis included cutaneous nodules, myopathy, macroglossia, skeletal pseudohypertrophy, or cutaneous purpura without other aetiology.
Authorisation for retrospective chart review was granted by the institutional review board of the Mayo Foundation in accordance with Minnesota state and federal regulations.
Results
We identified six patients with biopsy confirmed pulmonary amyloidosis and pulmonary marginal zone lymphoma. In five patients, diagnoses were made at the time of initial surgical histopathological review. One patient was referred to our institution for evaluation of localised pulmonary amyloidosis, and the diagnosis of concurrent pulmonary marginal zone lymphoma was made on review of the submitted histological sections of the lung specimen. All patients were followed for two years or until they died; no patient was lost to follow up.
CLINICAL CHARACTERISTICS
Table 1 summarises the clinical characteristics of our patients. All six patients were women. Age at time of diagnosis ranged between 45 and 85 years (median, 68). In symptomatic patients (four), presenting complaints included non-productive cough (two), generalised weakness and fatigue (one), and cough and weight loss (one). Examination findings were unremarkable. No adenopathy or organomegaly was noted.
Clinical characteristics
LABORATORY EVALUATION
Complete blood counts and serum chemistries were performed in all patients and were normal with the exception of patient 4, who was mildly anaemic.
Serum protein electrophoresis was performed in four of six patients (patients 1, 4, 5, and 6); no monoclonal spike was detected in these patients, but two patients (1 and 5) had polyclonal hypergammaglobulinaemia. Immunoelectrophoresis and immunofixation of the serum were performed in two patients (1 and 5) and were negative in both.
RADIOLOGICAL EVALUATION
All patients were evaluated initially with chest roentgenograms followed by chest computed tomography (CT) before diagnosis. In four of the six patients, multiple pulmonary nodules/masses were noted (fig 1). One patient had a single 6 cm pulmonary nodule. One had a single pulmonary nodule with diffuse interstitial infiltrates. Table 1 summarises the radiological findings.

Chest x ray demonstrating lung nodules in one patient. (A) Posteroanterior view of nodules; (B) lateral view of nodules.
BONE MARROW EVALUATION
Bone marrow aspiration and biopsy were performed in five of the six patients. All had normocellular bone marrows with normal haematopoietic precursors. No features of bone marrow involvement by malignant lymphoma, multiple myeloma, or amyloidosis were identified.
PULMONARY PATHOLOGY
Thoracotomy with single or multiple lobe resections was performed in all six patients. All pathology tissue specimens were obtained from lung parenchymal tissue and examined or reviewed at our institution. Extranodal marginal zone lymphoma was observed in the lung specimens from all six patients. In each case, the pulmonary parenchyma contained abnormal lymphocytic infiltrates. They formed masses that effaced the architecture of the lung and infiltrated the lung along the bronchovascular septa (fig 2). The infiltrates were composed of centrocyte-like cells with irregular nuclei, partially clumped chromatin, inconspicuous nucleoli, and moderately abundant pale staining to clear cytoplasm. In some areas, these cells infiltrated the pulmonary bronchiolar epithelium and formed lymphoepithelial complexes (fig 3). In addition, each lung specimen contained at least focally non-neoplastic germinal centres and clustered plasma cells. In the immunoperoxidase stains that were performed on the lymphomas (three cases), the neoplastic cells were positive for CD20 and the plasma cells exhibited immunoglobulin light chain restriction (κ in all three cases). The amyloid deposits were always easy to discern on routine haematoxylin and eosin stained sections. Amyloid either formed a large mass-like deposit accompanied by a granulomatous foreign body giant cell reaction or it infiltrated the walls of the pulmonary blood vessels (fig 4). In all cases, the amyloid was positive with the Congo red stain and showed apple green birefringence in polarised light. In addition, in three cases the amyloid stained positively for amyloid P component, and in all three it reacted positively with antibodies that recognise free κ immunoglobulin light chains and was negative for free λ immunoglobulin light chains. The immunoglobulin light chain type of the amyloid in these three cases matched the immunoglobulin light chain type expressed by the marginal zone lymphoma cells, including both the centrocyte-like cells and the intermixed clonal plasma cells. Tracheobronchial amyloidosis was not identified in these patients.
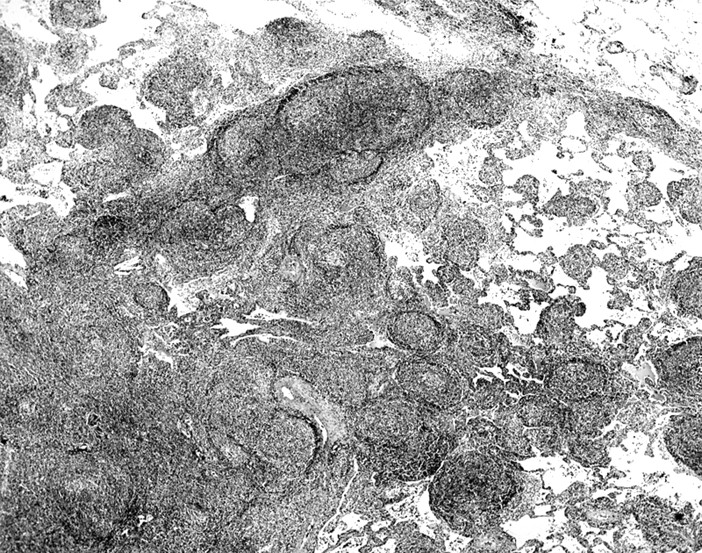
Extranodal marginal zone lymphoma involving the lung. Note the lymphocytic infiltrate effacing the pulmonary architecture and the spread of the abnormal lymphocytes along the bronchovascular septa (haematoxylin and eosin stained; orignal magnification, ×12).
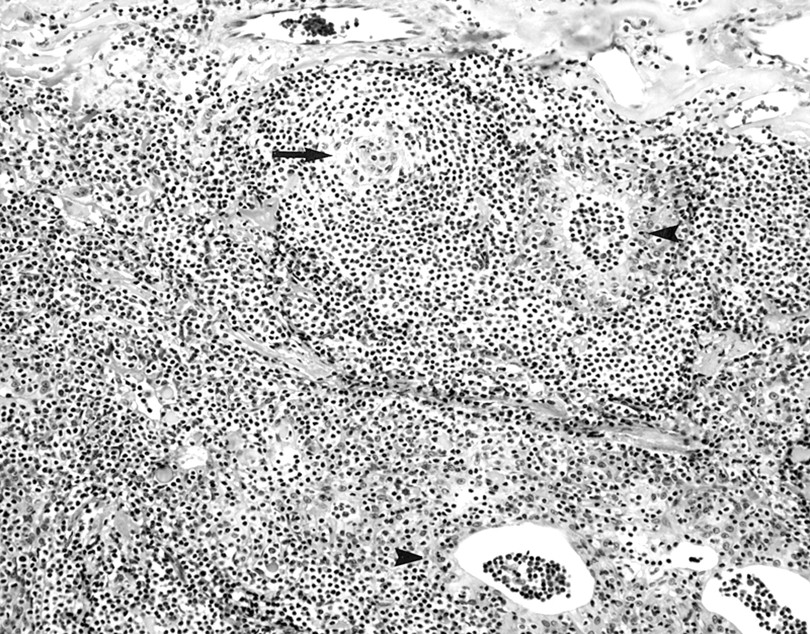
Extranodal marginal zone lymphoma involving the lung. The neoplasm is composed of centrocyte-like lymphocytes that infiltrate the bronchiolar epithelium and form lymphoepithelial complexes (arrowheads). An atrophic germinal centre is also present (arrow) (haematoxylin and eosin stained; original magnification, ×600).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pronounced amyloid deposition in a pulmonary extranodal marginal zone lymphoma. The amorphous material on the right side of the photograph is amyloid (haematoxylin and eosin stained; original magnification, ×600).
EVALUATION FOR AMYLOID SYNDROMES
Cardiac evaluation with electrocardiography was performed in all six patients. Four patients (patients 1, 4, 5, and 6) had normal findings, one patient (patient 3) had occasional premature ventricular contractions, and one patient (patient 2), with a known history of myocardial infarction, had an old anterior infarct. Echocardiography was performed in four of the six patients (patients 1, 2, 4, 5). All four had normal left ventricular size and function without evidence of overt congestive heart failure or diastolic dysfunction. Renal evaluation by urinalysis and creatinine was performed in all six patients. None had proteinuria or raised creatinine (median value, 7.6 mg/litre). Neurological evaluation by examination for upper or lower extremity neuropathy, autonomic neuropathy, carpal tunnel syndrome, or other focal neurological signs was performed in all patients. None had evidence of paresthesias, autonomic neuropathy, or focal neurological signs. Hepatic evaluation by physical examination and liver enzyme studies was performed in all patients. No hepatomegaly, splenomegaly, or liver enzyme abnormalities were noted. No reports or examination findings were seen that were consistent with cutaneous nodules, myopathy, macroglossia, skeletal pseudohypertrophy, or cutaneous purpura.
TREATMENT AND SURVIVAL
All six patients were managed initially by diagnostic and therapeutic surgical resection of known pulmonary nodules. Subsequent postsurgical management consisted of serial chest roentgenograms and CT scans, along with close clinical observation. Four of six patients are alive at survival durations of six, nine, three, and eight years (patients 1, 4, 5, and 6, respectively). One of the six patients (patient 2) died of unknown cause five years after diagnosis, at age 91.
One of the six patients (patient 3) died three years after diagnosis after she developed diffuse large B cell lymphoma.
One of the four surviving patients (patient 6) experienced recurrence of the primary disease in the soft tissue of the abdominal wall. After resection of two of three known pulmonary nodules in October 1990, the patient had an unremarkable course for less than one year. Follow up CT of the abdomen in May 1991 demonstrated increased density in subcutaneous tissue to the right of the umbilicus, which was suspicious for malignancy. The lesion slowly progressed until May 1997, when excision of the abdominal wall mass revealed malignant lymphoma, identical in morphology to previous lung specimens. She remains disease free at eight years.
Discussion
Lymphoma of MALT (extranodal marginal zone lymphoma) is a distinct type of extranodal B cell non-Hodgkin's lymphoma, which is characterised by a particular clinicopathological behaviour. First described in 1983 in a patient with primary gastrointestinal lymphoma15 and now subtyped as an extranodal marginal zone B cell lymphoma in the REAL classification, these lymphomas are composed of lymphocytes thought to be analogous to normal marginal zone lymphocytes, and typically recapitulate the cytology and architecture of normal Peyer's patches.16 They contain non-neoplastic germinal centres, and there is invasion of the epithelium, resulting in formation of the characteristic lymphoepithelial lesion.17 The gastrointestinal tract is most often involved (two thirds of cases), although the tumour has been seen with varying frequency in the lung, breast, liver, kidney, salivary gland, and skin.17
Localised amyloid deposition arising from extranodal marginal zone lymphoma is very rare. Caulet et al reported one patient in whom involvement of the small intestine and stomach with marginal zone lymphoma was found in association with Congo red and thioflavine T positive amyloidosis.10 Goteri et al reported four patients in whom gastrointestinal marginal zone lymphoma (three involving the stomach, one the large bowel) was associated with coexisting localised amyloid deposition.11 Shirasaka et al reported one patient in whom marginal zone lymphoma involving the stomach, lung, and submandibular gland was associated with localised amyloidosis of the colon and submandibular gland.12 Previous authors18, 19 have described cases of pulmonary small cell non-Hodgkin's lymphoma and extranodal lymphoplasmacytic immunocytoma in association with pulmonary amyloidosis. Because most cases of previously reported low grade B cell pulmonary lymphomas represent marginal zone lymphomas, it is likely that these authors were the first to describe localised amyloid deposits in association with this type of lymphoma. We report six cases in which this association can be made without evidence of coexistent systemic amyloidosis. In these patients the relative paucity of presenting symptoms and physical examination findings, and the absence of systemic amyloidosis after a rigorous clinical evaluation, corroborate the localised nature of the amyloid in pulmonary marginal zone lymphoma. In addition, in three cases we demonstrated that the amyloid was amyloid light chain type and that it matched the immunoglobulin light chain type expressed by the neoplastic marginal zone lymphoma cell. Therefore, it is likely that the abnormal immunoglobulin was produced by the neoplastic cells and was deposited locally in the tissues as light chain type amyloid.
Clinical presentation may be non-specific in both marginal zone lymphoma and pulmonary amyloidosis. Low grade extranodal marginal zone lymphomas and other low grade B cell non-Hodgkin's lymphomas are distinguished by their remarkable indolence—patients are often asymptomatic before diagnosis.20 Primary systemic amyloidosis, however, usually presents with discrete clinical manifestations (weakness, fatigue, weight loss, hepatomegaly, cutaneous purpura, ankle oedema, nephrotic syndrome, carpal tunnel syndrome, congestive heart failure, peripheral neuropathy, or orthostatic hypotension).2 Localised pulmonary amyloidosis is more often associated with few clinical symptoms, usually those related to the respiratory tract, such as productive or non-productive cough and dyspnea.21, 22 This was true in our series, in which five of six patients were either asymptomatic or had non-specific pulmonary or constitutional symptoms at the time of presentation.
Localised pulmonary amyloidosis remains an uncommon clinical presentation, representing 17 of 55 patients with pulmonary amyloid deposition seen at the Mayo Clinic between 1980 and 1993.2 Although the distribution of amyloid in the lungs and the association with primary disease and systemic illness have been well described,23 coexistent lymphoma has been reported in only two previous cases.18, 19 The observation of six patients with coexistent pulmonary marginal zone lymphoma and localised amyloidosis mandates further investigation into the pathogenetic mechanisms. In addition, the presence of amyloid deposits in the lung should raise suspicion for underlying or concurrent lymphoma.
Acknowledgments
Supported in part by research grant CA62242 from the National Institutes of Health.