Article Text

Abstract
Aims/Background—The advent of new treatments for haematological malignancies has led to the need for a correlation between cytogenetic and morphological abnormalities. This study aimed to achieve this by the application of interphase cytogenetics to marrow trephine sections, a technique not previously reported for formalin fixed, paraffin wax embedded trephine biopsies.
Methods—Dual colour fluorescence in situ hybridisation (FISH) was used to detect numerical and structural abnormalities in routinely processed paraffin wax embedded trephine biopsies. Three cases with t(8;21) and three with t(15;17) were analysed, together with a case of trisomy 8. Chromosome specific probes were hybridised with sections and disclosed by fluorescein isothiocyanate and rhodamine/Texas red labelled antidigoxigenin and antibiotin amplification; translocations were identified by colocalisation of probes using a double wavelength bypass filter.
Results—A translocation signal was present in 12% and 11.5% of the cells counted in the t(8;21) and t(15;17) cases, respectively, but in none of the normal controls (p < 0.001). In the case of trisomy 8, 9% of the cells counted contained three hybridisation signals for chromosome 8, whereas no cell contained more than two in the normal control (p < 0.001).
Conclusions—This technique is useful for archived routinely processed material, enabling it to be used as a research tool but also, and perhaps more importantly, in clinical practice.
- acute myeloid leukaemia
- paraffin wax embedded bone trephines
- cytogenetic abnormalities
- myelodysplasia
- fluorescence in situ hybridisation
Statistics from Altmetric.com
- acute myeloid leukaemia
- paraffin wax embedded bone trephines
- cytogenetic abnormalities
- myelodysplasia
- fluorescence in situ hybridisation
Precise localisation of cytogenetic abnormalities is not important for the diagnosis of acute myeloid leukaemia (AML), although the localisation of abnormal cells within the bone marrow is important both for the diagnosis and prognosis of myelodysplastic syndromes.1 It would also enable determination of the cytogenetic nature of abnormal cells after chemotherapy for AML, which may represent residual/recurrent disease, reactive or regenerating granulocytic cells, or secondary myelodysplasia. In addition, myelodysplasia is typified by the presence of abnormal localisation of immature precursors (ALIPs),2 which, although not exclusive to myelodysplasia (also found in reactive granulocytic hyperplasia), are prognostically important in cases with a low proportion of blasts.3 However, the cytogenetic nature of ALIPs is obscure4 and, particularly given their possible presence in reactive or neoplastic conditions, their characterisation at the molecular/cytogenetic level is important, both diagnostically and for an understanding of their biology. Interphase cytogenetics, specifically fluorescence in situ hybridisation (FISH), allows the cytogenetic and morphological features to be correlated,5 but has not been applied widely to routinely processed trephine biopsies, and the few studies that have used interphase cytogenetics in bone marrow trephines have all relied on single probes.6 Dual colour FISH using chromosome paints is superior to the use of single probes for the detection of translocations because it avoids the need to design and optimise new probes for each translocation,7 but this technique has not been applied to bone marrow trephines. We have developed dual colour FISH for the detection of translocations in interphase nuclei, in routinely processed bone marrow trephine biopsies, for use as a diagnostic and research tool.
Methods
CASE SELECTION
Archival formalin fixed, EDTA decalcified, paraffin wax embedded trephine biopsies were selected from the period 1991–9. Each had cytogenetic evidence, from contemporaneous metaphase spreads, of either t(8;21), t(15;17), or trisomy 8. Three cases of AML M2, with t(8;21) (age range, 25–43 years; one woman, two men); three of AML M3, with t(15;17) (age range, 22–59 years; three men); and one with MDS, and trisomy 8 (56 years, male) were used; four cytogenetically normal cases were used as controls.
SLIDE PREPARATION
Sections (7 μm thick) were mounted on coated slides, dried, and heated at 60°C for 48 hours. Slides were dewaxed and dehydrated before permeablisation with 100 μg/ml proteinase K (in 20mM Tris/HCl, 2mM CaCl2 (pH 7.4)) at 37°C for one hour. Proteinase K activity was stopped by dehydration and sections were treated with 1 mg/ml RNase A (Roche, Welwyn Garden City, UK) (in 10mM Tris (pH 7.5), 15mM NaCl) to reduce the background signal. Negative control sections were also treated with 10 U/100 μl DNase (in 40mM Tris/ HCl (pH 7.9), 10mM NaCl, 6mM MgCl2, 0.1mM CaCl2). DNase and RNase treatments were carried out for one hour at 37°C, followed by dehydration. Sections were denatured in 70% formamide, 2× saline sodium citrate (SSC) at 65°C for two minutes, followed by quenching in ice cold 70% ethanol for five minutes. Sections were dehydrated, air dried, and prewarmed to 37°C for two minutes before the addition of probe.
PROBE PREPARATION
Commercially available biotinylated chromosome specific paints for chromosomes 8 and 15 (Cambio, Cambridge, UK) and digoxigenin labelled chromosome 17 and 21 paints (Roche) were used. Biotinylated chromosome 8 paint was used for the identification of trisomy 8, whereas translocations were identified using differently labelled paints—biotinylated chromosome 8 paint and digoxigenin labelled chromosome 21 paints for t(8;21) and biotinylated chromosome 15 paint and digoxigenin labelled 17 paints for t(15;17). Biotinylated paint and hybridisation buffer were prewarmed to 42°C, mixed together (3 μl of paint + 12 μl hybridisation buffer/slide) and denatured at 65°C for 10 minutes, followed by one hour at 37°C. Digoxigenin labelled chromosome paints were supplied in hybridisation buffer; 10 μl/slide was denatured at 75°C for 10 minutes, followed by one hour at 37°C.
HYBRIDISATION AND POST-HYBRIDISATION STRINGENCY WASHES
Biotinylated chromosome paint (15 μl) and digoxigenin labelled chromosome paint (10 μl) were applied to translocation test slides, and 15 μl of biotinylated chromosome 8 paint was applied to trisomy test slides. An equivalent volume of hybridisation buffer without probe was added to negative control slides. The sections were incubated overnight at 37°C in a humid chamber. After hybridisation, coverslips were removed in 2× SSC and the slides were washed three times in 50% formamide, 2× SSC, once in 2× SCC, and once in 2× SSC, 1% IGEPAL CA-630 (Sigma, Poole, Dorset, UK), each for five minutes at 45°C. This was then followed by two washes at room temperature for five minutes in PN buffer (100mM sodium phosphate (pH 8.0), 0.1% IGEPAL CA-630).
SIGNAL DETECTION
Non-specific signal was reduced by a 30 minute incubation in a 0.5% solution of Roche blocking reagent (Roche) in a solution of PN buffer at 37°C, followed by two five minute washes in PN buffer. All incubations and washes were performed in the dark to minimise background. Biotinylated probes were detected by a 20 minute incubation at 37°C with 10 μg/ml avidin–fluorescein isothiocyanate (FITC; Sigma) in PMN buffer (100mM sodium phosphate, 0.1% IGEPAL CA-630, 0.02% sodium azide (Sigma), 5% bovine serum albumin (Sigma)), followed by washing twice in PN buffer for five minutes. The FITC signal was amplified by a 20 minute incubation at 37°C with biotinylated anti-avidin (Sigma; 1/1500 dilution) in PMN buffer, followed by two washes in PN buffer and incubation at 37°C for 20 minutes in 10 μg/ml avidin–FITC in PMN buffer. Digoxigenin labelled probes were detected by rhodamine conjugated sheep antidigoxigenin (Roche) (33 μg/ml) in PMN buffer, followed by washing in PN buffer, and the signal amplified by incubation at 37°C in donkey antisheep Texas red conjugated antibody (Jackson Immuno Research, West Grove, USA; 75 μg/ml) in PMN buffer. Slides were washed in PN buffer and counterstained with DAPI (Sigma; 2 μg/ml for 10 minutes at 37°C). The slides were then washed in PN buffer and phosphate buffered saline, mounted in 25% DABCO/glycerol (Sigma), covered, and sealed.
IMAGE ANALYSIS
Slides were visualised under fluorescent light using a double wavelength bypass filter (Leica, Milton Keynes, UK) and images directly captured photographically. Translocation signals appeared yellow owing to the colocalisation of the two probes to the translocation site. The number of cells containing one, two, three, or four single colour hybridisation signals and the number containing a translocation signal (in the translocation cases), or three signals of the same colour (in the trisomy cases) was recorded for each case; signals were only counted in non-overlapping cells. The incidence of each signal type was compared between tests and controls using the χ2 test.
Results
TRISOMY 8
Cells containing three signals were present in 9.7% of cells in the case of trisomy 8 (fig 1A), but not in the normal control case (p < 0.001); 400 and 200 cells were inspected in the trisomy 8 case and the normal control, respectively. One and two signals were present in 63.8% and 26.5% of cells in the trisomy 8 case and in 82.8% and 17.2% of cells in the normal control, respectively; negative control sections contained no hybridisation signal (fig 1).

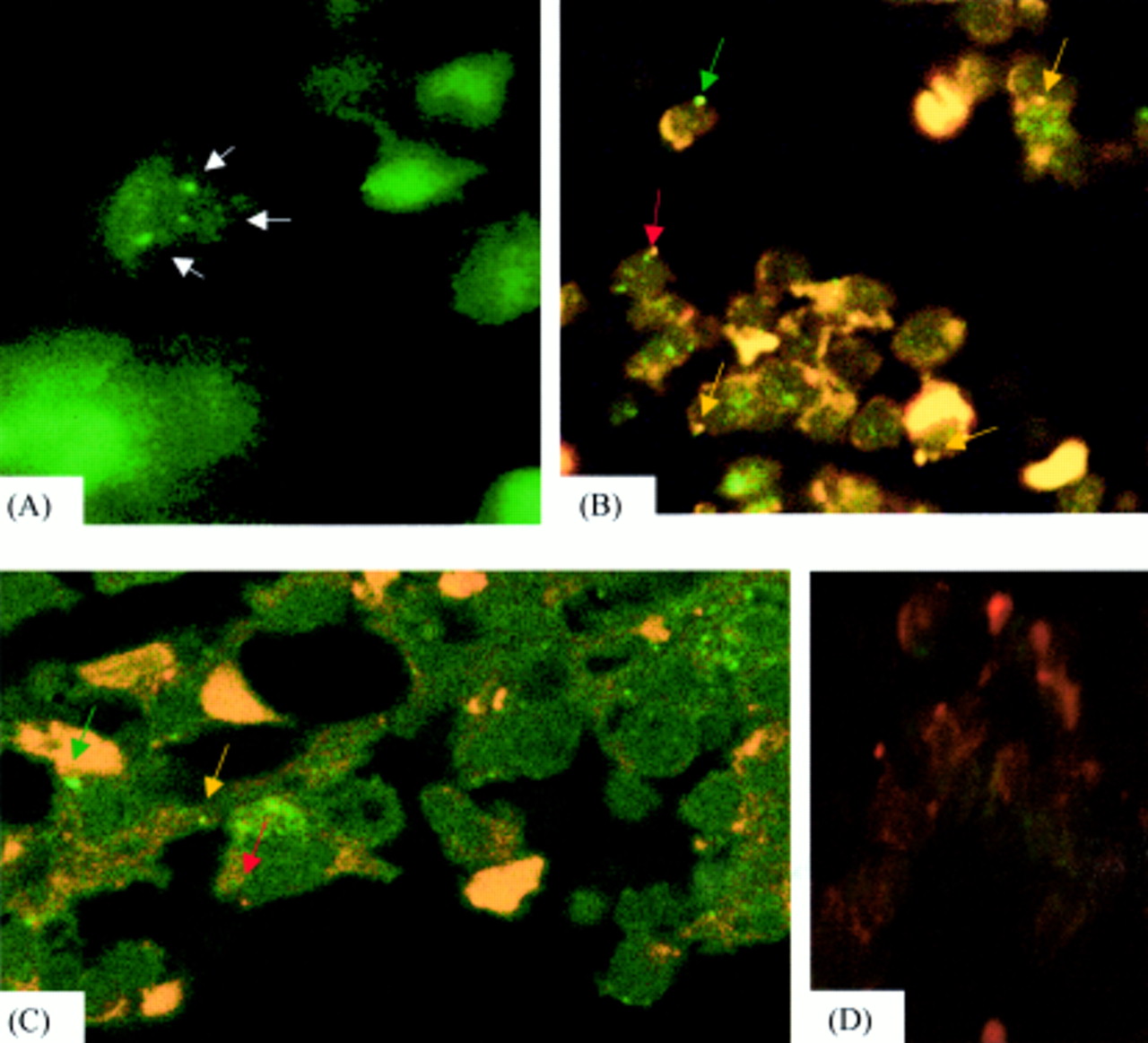{kind=link}
(A) Hybridisation for chromosome 8 paint, demonstrating three signals in the case of trisomy 8 (arrows). (B) Yellow hybridisation signal of t(8;21) as a result of colocalisation of red and green chromosome 8 and 21 paints (colour coded arrows). (C) Yellow hybridisation signal in a case of t(15;17) as a result of colocalisation of red and green chromosome 15 and 17 paints (colour coded arrows). (D) No hybridisation signal was seen in DNase negative controls, although some non-specific autofluorescence was present.
TRANSLOCATIONS
For both t(8;21)(q22;q22) and t(15;17) (q22;q11), yellow translocation signals were present in all cases with a translocation, but in none of the normal control cases (fig 1B,C). For t(8;21), translocation signals were present in a mean of 12% (range, 9.7–14%) of the 400 cells inspected (p < 9.6 × 10−12), whereas for the t(15;17) cases, a mean of 11.5% (range, 6–27%) of the 700 cells inspected contained a translocation signal (p < 1.03 × 10−7); negative control sections contained no hybridisation signals.
Discussion
Interphase cytogenetics using FISH allows the direct correlation of cytogenetic and morphological features in tissue sections, but has not been applied widely to bone marrow trephine biopsies. Thiele and colleagues6 used single probe FISH to localise the Philadelphia chromosome in chronic myeloid leukaemia (in biopsies), whereas Nolte and colleagues8 used dual colour FISH for bcr and abl, in 13 cases, but to date there is no report using chromosome paints, which are inherently more flexible in application, in trephine sections.
Because the nuclei of the cells were sectioned, only a proportion will contain either a translocation signal or evidence of trisomy. To minimise this effect, we used 7 μm sections; thicker sections would necessitate the use of confocal microscopy. The proportions of cells containing a translocation signal or three chromosome 8 signals were similar to those reported for studies using whole cell preparations. Wilkens and colleagues9 found chromosome 8 signals in between 0% and 3% of cells in normal controls, on the basis of which the presence of three hybridisation signals in more than 3% of cells was used as the cut off point for the determination of true trisomy. Interestingly, no cells containing three signals were seen in the control case, which is in agreement with previous reports. Furthermore, although some studies have found more cells containing two signals than one, these were based on the study of whole nuclei. In agreement with our findings, Southern and Herrington10 found the reverse for hybridisation to 7 μm thick tissue sections. Similarly, the lack of translocation signals in control sections, which can occur in whole cell preparations as a result of overlapping, was absent in our study. This is because, as a result of tissue sectioning, hybridisation occurred in a single plane, precluding the possibility of overlap.
The transfer of the technique to formalin fixed, decalcified, paraffin wax embedded bone marrow trephine sections renders it applicable to diagnostic practice, for differentiation of residual disease and regenerative changes after chemotherapy. Furthermore, because it was successful in archival material nearly 10 years old, it can be used on stored trephines to study the nature and role of ALIPs in progression to acute myeloid leukaemia.
Acknowledgments
The generous support of the Peel Medical Trust is gratefully acknowledged.