Article Text

Abstract
This paper describes the case of a 48 year old man who presented with acute hyperlipidaemia following pulmonary embolism. Subsequent investigation revealed that the hyperlipidaemia was secondary to nephrotic syndrome of glomerulonephritis. The case illustrates the importance of investigating acute hyperlipidaemia for its underlying causes.
- membranous glomerulonephritis
- pulmonary embolism
- acute hyperlipidaemia
- nephrotic syndrome
- HDL, high density lipoprotein
- IDL, intermediate density lipoprotein
- LDL, low density lipoprotein
- NSF-CHD, National Service Framework for Coronary Heart Disease
- VLDL, very low density lipoprotein
Statistics from Altmetric.com
- HDL, high density lipoprotein
- IDL, intermediate density lipoprotein
- LDL, low density lipoprotein
- NSF-CHD, National Service Framework for Coronary Heart Disease
- VLDL, very low density lipoprotein
A 48 year old man presented to the admissions unit with acute onset right sided pleuritic chest pain and dyspnoea. On examination he was apyrexial and tachycardic (100 beats/minute sinus rhythm). Crepitations were heard at the right base. Subsequent ventilation/perfusion scanning revealed an unmatched perfusion defect on the right side, consistent with a pulmonary embolism. He was anticoagulated and discharged home. During his hospital admission his total cholesterol concentration had been measured as 7.5 mmol/litre.
Three weeks later he consulted his general practitioner complaining of tiredness and weight gain (he estimated nearly two stone over two months). Investigation revealed a total cholesterol of 14.4 mmol/litre, triglyceride of 4.87 mmol/litre, and high density lipoprotein (HDL) of 1.18 mmol/litre. Total protein concentration was 51 g/litre, albumin was 22 g/litre, and γ glutamyl transpeptidase was 77 U/litre. Other liver function tests were normal. The patient had previously attended regular annual well man clinics, where his highest recorded total cholesterol concentration was 5.5 mmol/litre. In view of his hyperlipidaemia his general practitioner considered starting him on statin treatment and contacted us to seek our advice regarding the most appropriate agent to treat such high lipid concentrations. After discussion of the acute onset of the hyperlipidaemia and the abnormal liver function tests, the patient was referred to the lipid clinic for further investigation.
He attended one week later. He continued to complain of worsening fatigue. He also reported intermittent bilateral loin and suprapubic pain of recent onset (three to four weeks). Over the same period he had noticed nocturia and increasing urinary frequency. He had recently stopped smoking, having previously smoked 30 cigarettes daily for about seven years. His alcohol consumption was approximately 6 units/week.
He weighed 96.5 kg and had a body mass index of 34.2 kg/m2. His blood pressure was 160/100 mm Hg. There was mild peripheral oedema. Examination of the chest was normal. Abdominal examination revealed some loin tenderness but no other specific abnormality.
Serum lipid concentrations and liver function tests were repeated and were consistent with results from the previous week. Further investigations revealed microscopic haematuria and considerable proteinuria (urinary albumin : creatinine ratio, 398 mg/mmol; normal range, < 3.5; 24 hour urinary albumin excretion, 4514 mg). Serum urea and electrolyte concentrations were normal. Fasting plasma glucose was 5.4 mmol/litre. Antineutrophil cytoplasmic antibodies, antiperoxidase 3 antibodies, myeloperoxidase, anti-glomerular basement membrane, and antinuclear factor antibodies were negative. C3 and C4 values were not raised. Serum IgA was slightly raised at 5.07 g/litre, with a polyclonal pattern on electrophoresis. Serum IgG was low at 3.7 g/litre.
Abdominal ultrasound was unremarkable. A renal biopsy was arranged.
DIFFERENTIAL DIAGNOSIS
The acute nature of the dyslipidaemia is indicative of a secondary hyperlipidaemia, which usually results from drug treatment or another disease state (table 1). The management of these cases is directed towards the identification and treatment of the primary condition, with the secondary lipid disturbances often responding as the underlying stimulus to hyperlipidaemia is removed.1 It is good practice to assess the need for specific lipid lowering medication after treatment of the primary disorder. Inappropriate initiation of lipid lowering medication may delay appropriate investigation of the underlying cause and even add to the burden of morbidity as a result of potential side effects.2,3
Common causes of secondary hyperlipidaemia
In our case, the triad of hypoalbuminaemia, heavy proteinuria, and oedema clinically identifies nephrotic syndrome as the cause of the acute hyperlipidaemia. The glomerulonephritides are the most common cause of nephrotic syndrome, others being systemic vasculitis, diabetic glomerulosclerosis, and drugs (such as penicillamine, gold, and captopril).4
The pattern of dyslipidaemia seen in our patient is typical of that observed in advanced nephrotic syndrome with raised total cholesterol, raised triglycerides, and low, normal, or occasionally even slightly raised HDL.5 The magnitude of the lipid abnormality correlates with disease severity, but neither the pattern nor the degree of the dyslipidaemia gives any indication as to the nature of the underlying glomerular pathology.6 The raised serum total cholesterol concentration is predominantly the result of an increase in low density lipoprotein (LDL), with very low density lipoprotein (VLDL) concentrations also raised in some cases. Reduced lecithin: cholesterol acyltransferase activity results in a reduction in HDL2 and an increase in HDL3 subfractions.5
There is evidence that the mechanism underlying these dyslipidaemic changes is one of enhanced hepatic LDL production.7,8 The trigger appears to be a decrease in oncotic pressure, rather than simple hypoalbuminaemia, because infusions of either albumin or dextran will normalise lipoprotein concentrations.9 Low oncotic pressure has been shown to stimulate hepatic apolipoprotein B gene transcription in vitro.10
The hypertriglyceridaemia of nephrotic syndrome is not the result of increased synthesis, but rather disturbed catabolism. The delipidation process in which VLDL is converted to intermediate density lipoprotein (IDL) and then to LDL by lipoprotein lipase is impaired.11 LDL receptor clearance of both LDL and IDL is also reduced.11,12
Abnormalities of coagulation are well recognised in nephrotic syndrome.13,14 By far their most common clinical manifestation is thromboembolism, although bleeding tendencies have been reported in some patients.14 This helps explain the patient's initial presentation with pulmonary embolism. The mechanism underlying the thrombotic tendency has not been fully elucidated, although many relevant processes correlate with clinical episodes. Dehydration, with subsequent haemoconcentration and hypovolaemia, is frequently seen, with a resultant increase in whole blood and plasma viscosity.14,15 Hypoalbuminaemia stimulates hepatic synthesis of several coagulation factors including factors V, VII, X, and XIII, fibrinogen, and fibronectin.14,16–18 The plasma protein profile in nephrotic syndrome classically shows low antithrombin III and free protein S, whereas protein C values are normal or raised.13,19 Urinary losses of antithrombin III and protein S are pronounced in patients with nephrotic syndrome.20 Although antithrombin III values are low, total activity is normal because of a rise in α macroglobulin concentration. However, the loss of protein S contributes greatly to thrombotic tendency. Hypoalbuminaemia reduces plasminogen binding to fibrin, suppressing fibrinolysis.21 It also results in greater availability of arachidonic acid for the synthesis of proaggregant thromboxane A2 within platelets.22 Platelet activation and aggregability is further stimulated by high plasma concentrations of cholesterol, fibrinogen, and von Willibrand factor.14,23 The severity of the haemostatic disorder generally parallels the degree of albuminuria and hypoalbuminaemia.13 The increasing immobility forced on patients by the complications of nephrosis is an additional factor favouring thrombosis.
The low IgG concentration is a reflection of non-selective urinary protein loss. The slightly raised IgA value remains unexplained, although no other respiratory or gastrointestinal pathology is evident.
CLINICAL DIAGNOSIS: PULMONARY EMBOLISM AND ACUTE HYPERLIPIDAEMIA SECONDARY TO NEPHROTIC SYNDROME
Pathological diagnosis
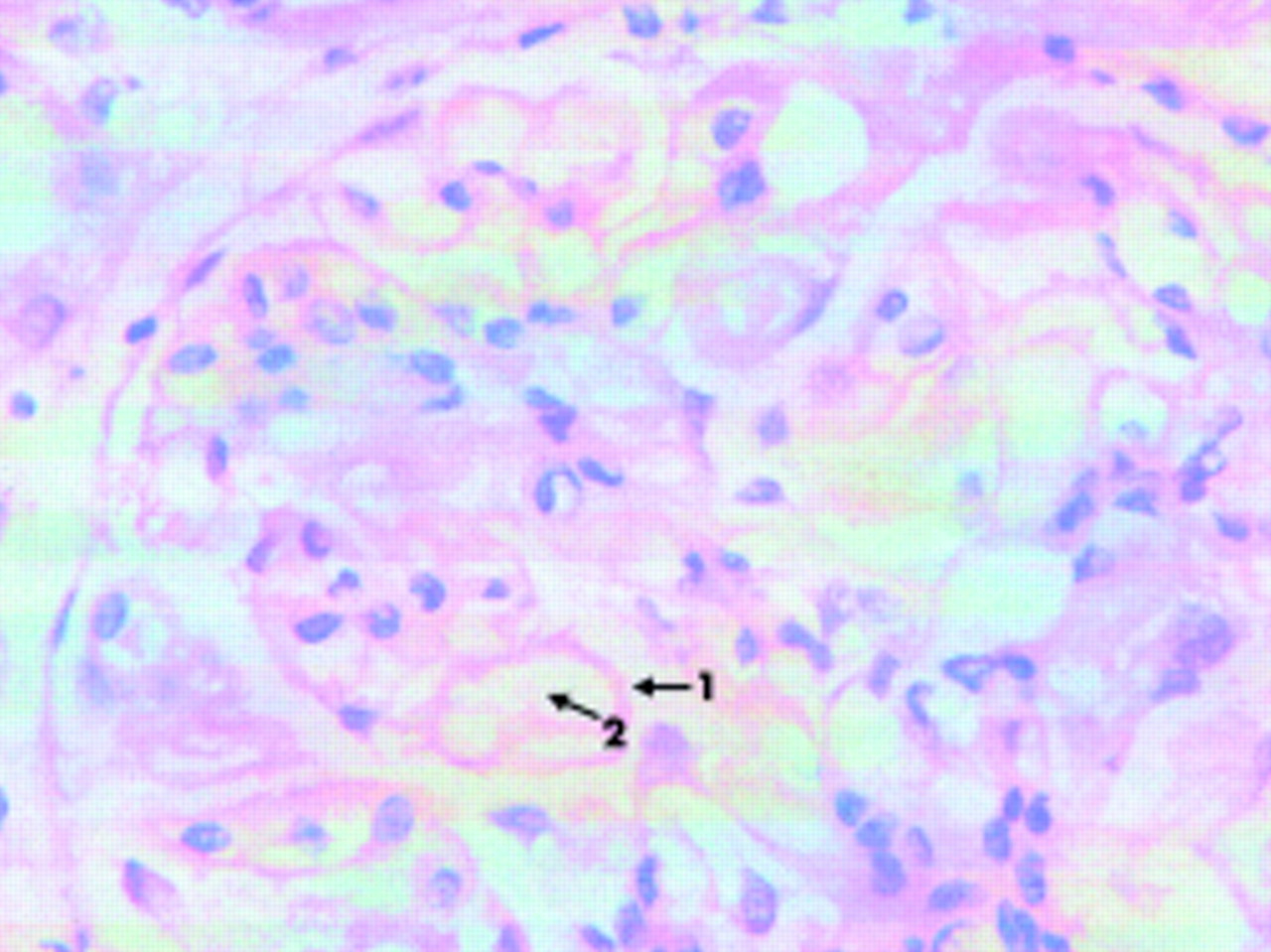
The renal biopsy showed glomeruli that had mild thickening of the capillary walls with patent capillary loops. There was no mesangial hypercellularity and the tubules, interstitium, and vessels were normal. Immunofluorescence showed strong granular IgG along the capillary wall and some complement. No deposition of IgA was seen. The features were those of membranous glomerulonephritis (fig 1).

{kind=link}
Microscopic picture of renal biopsy showing thickened glomerular walls and patent capillary loops. Arrow 1, thickened capillary wall; arrow 2, patent capillary loop.
Pathological diagnosis
Membranous glomerulonephritis.
DISCUSSION
Membranous glomerulonephritis can be primary (idiopathic) or secondary to drugs (such as non-steroidal anti-inflammatory drugs, gold, penicillamine), malignancy (breast or bronchial carcinoma), infection, or hypothyroidism.14 No secondary cause has been found in this case.
In addition to anticoagulation, the patient was started on frusemide before a pathological diagnosis was obtained. This was for symptomatic relief of his oedema, which had probably been masked to some extent by pre-morbid obesity. Membranous glomerulonephritis is treated with immunosuppressive treatment. At least 25% of cases resolve spontaneously, with treatment reserved for those with deteriorating renal function or severe nephrotic syndrome. Progressive renal failure occurs in approximately 30% of patients, despite immunosuppression.24 In these cases, long term nephrotic syndrome and its associated lipid disturbance presents a high risk for the development of atherosclerotic disease. The potential benefits of lipid reduction warrant the cautious use of lipid lowering medication, in addition to the introduction of a low saturated fat diet.25,26 Fibrates accumulate in renal disease and are associated with an increased risk of myotoxicity in these patients.2,3 Statin treatment, usually at low dose, is the treatment of choice.27 Cyclosporin A increases the myotoxic tendency of some statins, necessitating regular clinical and biochemical monitoring of patients managed on this treatment combination.2
Take home messages
-
Acute hyperlipidaemia is always an indication for further investigation
-
Baseline investigations must be performed to exclude secondary causes of hyperlipidaemia before starting a patient on lipid lowering medication
Our patient was initially treated with cyclosporin A for three months. This failed to resolve his nephrotic syndrome. In addition, he experienced side effects in the form of nausea and vomiting. His anticoagulation was also disturbed. Cyclosporin has now been discontinued. His renal function is slowly deteriorating despite the treatment outlined. He is beginning treatment with low dose statin.
The presentation of this case is timely given the recently published National Service Framework for Coronary Heart Disease (NSF-CHD)28 because it illustrates a potential future problem in the management of lipid disorders. The NSF-CHD places the emphasis for the diagnosis and treatment of these on to primary care. In its white paper Saving Lives: Our Healthier Nation 1999,29 the Government pledged to reduce the death rate from heart disease and stroke in the under 75s by two fifths over 10 years. Subsequently, in the NSF-CHD published in March 2000, primary care is given a responsibility for maintaining secondary prevention in patients with established coronary heart disease (Standard 3) and for implementing primary prevention for individuals identified as being at high future coronary heart disease risk (Standard 4).28 Control of cholesterol by statins is an important component of this strategy for high risk patients, and the NSF contains target cholesterol values for such individuals.
Among the mechanisms suggested by the NSF for the systematic delivery of such care are nurse run “cardiac prevention clinics”. It is possible that in the future prescription of statins by nurses may be allowed under group directive protocols.
Increasing numbers of patients are having their cholesterol measured on a regular basis. Most of these requests already originate from primary care, as do the management decisions based on the results. For instance, in Walsall during 2000/2001, 71% of all requests for cholesterol measurement and 91% of requests for fasting lipids originated in primary care.
It is important that the possibility of secondary hyperlipidemia should be excluded in all patients considered for lipid lowering treatment. Although the causes of secondary hyperlipidaemia are well known to chemical pathologists and metabolic physicians, this knowledge may not be shared by many general practitioners, and still less so by primary care nurses, who encounter these conditions less frequently. The inappropriate initiation of lipid lowering medication in such cases, in addition to delaying appropriate investigation of the underlying causes, may be dangerous in itself. Lipid lowering agents are not without side effects, the risk of which is increased by nephrotic syndrome and renal failure, recognised causes of secondary hyperlipidaemia.2 It is recommended that common causes of secondary hyperlipidaemia should be excluded by a biochemical screen of urinalysis for protein, fasting plasma glucose concentration, thyroid function tests and liver function tests before starting a patient on lipid lowering drugs.30,31 It is important that those hospital clinicians with a special interest in lipid disorders should take a lead educational role in making primary care teams aware of these issues.