Article Text

Abstract
Background Mutation detection in tumours will become increasingly important in pathological diagnosis as ‘predictive’ mutations are identified. A cheap and reliable test that works on formalin-fixed paraffin-embedded (FFPE) tissue is required.
Methods The quick-multiplex-consensus (QMC)-PCR protocol was developed to be used with high-resolution melting (HRM) analysis. The assay was compared with Sanger sequencing. Robustness of the assay was tested in DNA from FFPE tissue.
Results QMC-PCR with HRM could detect a minimum of 2.5% of mutant alleles (compared with 20% detectable for Sanger sequencing). Ten mutation hotspots in KRAS, BRAF, PIK3CA and CDC4 were screened in 29 cell lines with 100% sensitivity and specificity. Forty-three FFPE colorectal tumours were sequenced for hotspots in KRAS and PIK3CA and then screened by QMC-PCR. There was 100% sensitivity, although, of 21 mutations detected by QMC-PCR, 16 were confirmed by sequencing (71% specificity, positive predictive value 76%). All 43 samples were then screened for mutations in all 10 hotspots. Of 430 tests, 43 (10%) showed aberrant melting and 36 were confirmed mutant (positive predictive value 84%). As our technique is more sensitive than direct sequencing, the remaining seven tests are probably sequencing false-negatives. Precision tests showed that there was little intra-assay and interassay variation.
Conclusions QMC-PCR with HRM is a simple, robust and inexpensive technique which had greater sensitivity than Sanger sequencing. It allows multiple mutation hotspots to be rapidly screened and is thus highly suited to mutation detection in DNA derived from FFPE tissues.
- PCR
- HRM
- mutation detection
- formalin-fixed paraffin-embedded
- cancer genetics
- DNA
- molecular pathology
Statistics from Altmetric.com
Introduction
Cancer management has traditionally depended heavily on histopathology. From morphological evaluation of the initial diagnostic biopsy to mapping of disease spread in resection specimens, the pathologist has been central to the decision-making process in most types of tumour. Descriptive analysis of tumours is, however, prone to interobserver variation and does not give any insight into tumour biology.1 As the number of treatment options increases, new criteria—in addition to those used by pathologists—are required for more consistent classification/staging of tumours and to allow bespoke tailoring of therapies based on tumour biology. New criteria may comprise ‘profiling’ the mutations present within a tumour. Some mutations are specific for certain types and subtypes of cancer and can therefore be used for diagnostic classification of those cancers.2 Other mutations seem to carry prognostic importance, and, perhaps most importantly, some mutations are predictive of response to specific therapies.3–6 For example, it has been shown that the presence of KRAS mutations in colorectal cancers abrogates tumour response to anti-epidermal growth factor receptor therapies, and the National Comprehensive Cancer Network guidelines now recommend routine testing for KRAS mutation in all cases where such therapy is being considered (http://www.nccn.org/).
Predictive testing assumes even more importance when neoadjuvant chemotherapies are being considered, because significant clinical down-staging can be achieved before definitive surgery, and thus correct choice of therapy is essential. A variety of techniques can be used for mutation analysis. Direct DNA sequencing is generally regarded as the ‘gold standard’, although it is expensive, laborious and gives false-negative results when a large number of normal cells are admixed with tumour cells. As mutations can occur in multiple sites in a gene (usually known as ‘hotspots’) and multiple genes may be mutated within a specific pathway, detection of pathway disruption by direct sequencing would be prohibitively expensive. Screening of hotspots for mutation before sequencing could reduce this cost by excluding wild-type samples from further analysis. Techniques such as single-strand conformation polymorphism,7 temperature gradient gel electrophoresis,8 denaturing high-performance liquid chromatography9 and matrix-assisted laser desorption/ionisation-time of flight mass spectroscopy10 11 have been developed for the purposes of mutation detection. However, these techniques are complicated, expensive and tend to be available only in research institutions. Furthermore, even in research institutes, routine practice is for tumour tissue to be formalin-fixed and paraffin-embedded (FFPE) to allow histological analysis. Tissues thus processed tend to suffer DNA degradation, and the utility of sophisticated screening techniques on this type of template remains to be proven.
High-resolution melting (HRM) analysis is a cheap in-tube (ie, non-gel-based) mutation detection technique that works on the basis that a difference in DNA sequence results in differences in the way the DNA melts.12 This is particularly pronounced when mutant and wild-type alleles are mixed, as the formation of heteroduplexes results in a unique melting pattern. Unlike the other techniques, it requires no post-PCR processing, expensive fluorescent-labelled oligonucleotides or allele-specific PCR. Interpretation of the data is very simple. HRM was initially developed for detection of single-nucleotide polymorphisms (SNPs), but it is suitable for mutation analysis.13 The HRM technique has been used to identify somatic mutations in a variety of genes,12 and recently it has been used for quantitative methylation detection.14
As with all the screening techniques, HRM is dependent on the quality of the DNA template. Our experience hitherto and that of others is that HRM on FFPE tissue is prone to false-positive artefacts.15 This study sought to refine the HRM technique to make it sufficiently robust for diagnostic use with FFPE tissue. We describe a simple, standardised and rapid methodology that allows multiple genes to be screened and analysed in an automated fashion. With the use of this technique, a mutation profile of a tumour can be rapidly produced. Furthermore, it is sufficiently cheap and simple to be implemented in pathology laboratories in both teaching hospitals and non-teaching hospitals.
Materials and methods
DNA template
For initial optimisation of PCR protocols, high-quality DNA was obtained from 29 well-described cell lines as previously described.16 Validation of PCR protocols was undertaken on DNA extracted from FFPE tissue, as this is representative of material usually available in the diagnostic setting. Ethics approval was obtained for the use of patient materials in this study (reference No C02.310). Tumour blocks from 50 colorectal cancers from 37 patients who underwent surgery at the Queen's Medical Centre (between 1983 and 2005) were retrieved. Six sections (each of 10 μm thickness) were cut from each block and DNA extracted using the QIAamp DNA FFPE tissue kit (Qiagen, Crawley, UK; Cadama Medical Ltd, Stourbridge, UK; PeQLab Biotechnology, Fareham, UK; Idaho Technology, Salt Lake City, USA; Roche, Welwyn Garden City, UK) following the manufacturer's protocol.
Sample quality control
All DNA samples were tested for quality using three pairs of primers for GAPDH in a multiplex PCR to give products of 100, 200, 300 and 400 bp.17 PCR was performed in a 10 μl reaction containing 1×HotShot master mix (Cadama Medical Ltd), each primer at 0.250 μM final concentration and 20 ng template DNA. A Primus96 advanced Gradient Thermal Cycler (PeQLab Biotechnology) was used with the following protocol: (95°C/5 min) ×1; ((95°C/30 s)/(62°C/1 min)/(72°C/1 min)) ×45; (72°C/10 min) ×1. PCR products were detected by adding 1× LCGreen PLUS (Idaho Technology) and undertaking HRM analysis as described below.
Quick-multiplex-consensus (QMC)-PCR protocols
Primer design
QMC-PCR is a nested procedure in which an initial pre-diagnostic multiplex (PDM) reaction is followed by a single specific diagnostic (SSD) reaction. The PDM reaction contained outer primer pairs for up to 10 different target sequences in a single reaction. The SSD reaction contained just a single inner primer pair specific to each target. All primers were designed using Primer 3 software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi); for the PDM reaction, primers were tested against the others by in silico PCR (http://genome.csdb.cn/cgi-bin/hgPcr/), and only non-cross-reacting primers were selected. Primers covered hotspots in KRAS (exons 2, 3 and 4 encompassing codons 12/13, 61 and 146, respectively), BRAF (exons 11 and 15 (the latter encompassing codon 600)), PI3KCA (exons 1, 9 and 20) and CDC4 (exons 8 and 10). The nature of HRM and the problems engendered when working with DNA from FFPE tissues does impose some limits. Primers were designed to ensure a maximum PCR product size of around 200 bp. Primer sequences are given in supplementary table 1 (available online only).
Thermal cycling conditions
All PCRs were performed on the thermal cycler described above. The PDM reaction was undertaken in a final volume of 25 μl. Each reaction contained 1×HotShot master mix, 10 primer pairs (covering all the above hotspots) with each primer at 0.400 μM final concentration and 20 ng template DNA. PCR was performed using a two-step protocol: (95°C/5 min) ×1; ((95°C/1 s)/(55°C/1 s)) ×25. The total duration was 38 min.
The SSD reaction was in a final volume of 10 μl, which contained 1×HotShot master mix, 1 primer pair with each primer at 0.250 μM final concentration and 1× LC Green PLUS. The template consisted of 1 μl of a 1:100 dilution of the product from the PDM reaction, and PCR was performed using a two-step protocol: (95°C/5 min) ×1; ((95°C/1 s)/(55°C/1 s)) ×45. The total duration was 58 min.
Cold PCR protocols
For comparison with QMC-PCR, we also tested ‘cold’ PCR protocols that have recently been described as modifications of the standard PCR protocol for enriching mutant alleles.15 The ‘fast cold’ PCR protocol basically reduces the temperature for denaturation to a specific critical temperature (Tc), but will only enrich for certain mutation types. The ‘full cold’ PCR enriches for all mutations, but introduces two extra steps to the standard three-step protocol (ie, hybridisation at 70°C and denaturation at Tc) and lasts over 4 h. The Tc needs to be determined empirically for each exon and both fast and full cold protocols were tested (see online supplementary data).
HRM analysis
The PCR products of the SSD reaction were transferred to Light Cycler capillaries (20 μl) (Roche). The products were melted in the HR-1 HRM instrument (Idaho Technology) at a rate of 0.3°C/s, with a starting temperature of 60°C, a final temperature of 90°C and fluorescence data acquisition at 70–85°C. The data were analysed using the HR-1 analysis tool custom software, and both derivative plots and difference plots were generated after normalising and temperature shifting. The derivative and difference plots were visually inspected in order to separate out the mutants from the wild-type samples using a threshold of 4% difference in fluorescence as previously described.16 18
Assay performance
Evaluation in cell lines and DNA from FFPE tissue
DNA from cell lines was used to initially optimise the cycling conditions for QMC-PCR. In order to test the assay in the cell lines against the ‘gold standard’ of sequencing, 29 cell lines underwent sequence analysis for the hotspots in KRAS and BRAF, and these were then screened by QMC-PCR. Subsequently, 29 cell lines underwent screening for all 10 hotspots, and those noted as ‘mutant’ were validated by direct sequencing.
To test the assay in DNA derived from FFPE tissue, 43 samples underwent direct sequencing analysis for the hotspots in KRAS exon 2 and PIK3CA exon 20 and were then screened by QMC-PCR. Subsequently, the 43 samples were screened for all 10 hotspots, and those noted as ‘mutant’ were validated by direct sequencing.
Limit of detection
To assess the limit of detection of QMC-PCR, we performed dilution experiments by spiking DNA from diploid cell lines with known mutations into diploid cell lines known to be wild-type at that hotspot. Thus DLD1 (containing a heterozygous E545K mutation in PIK3CA) was spiked into VACO5 (wild-type at codon 545). Similarly VACO5 (heterozygous V600E mutant in BRAF) was spiked into HCT116 (wild-type in BRAF). Spiking was carried out to produce 40%, 20%, 10%, 5%, 2.5% and 1.25% mutant alleles in the DNA mix.
Comparison with ‘standard’ PCR/HRM in DNA derived from FFPE tissue
The QMC-PCR protocol was also compared with a standard protocol for the hotspot in BRAF exon 15 using DNA derived from FFPE tissue. For the standard reaction, the mixture contained 1×HotShot master mix, primers at 0.250 μM concentration, 1× LCGreen PLUS with 20 ng template. PCR was performed using the following cycling conditions: (95°C/5 min) ×1; ((95°C/30 s)/(55°C/1 min)/(72°C/1 min)) ×45. The total duration time was 2 h and 55 min, and the PCR products then underwent HRM analysis as described above.
Short-term and long-term precision in DNA derived from FFPE tissue
Intra-assay and interassay variation (respectively short-term and long-term precision) was also assessed. For the intra-assay variation, tests were conducted for exon 20 of PIK3CA and in exon 15 of BRAF. Two FFPE-derived DNA samples, one wild-type and one mutant, were tested as 15 replicates for each sample on two separate occasions. To test interassay variation, a series of six FFPE-derived samples (three mutant and three wild-type for exon 20 of PIK3CA) were tested on 3 consecutive days.
Sequencing
Tumour samples showing abnormal HRM patterns suggesting mutation were sequenced using the appropriate PCR primers as previously described.16 The chromatograms were viewed and interpreted using the Chromas Lite software V2.01.
Results
Quality control and single-step PCR/HRM
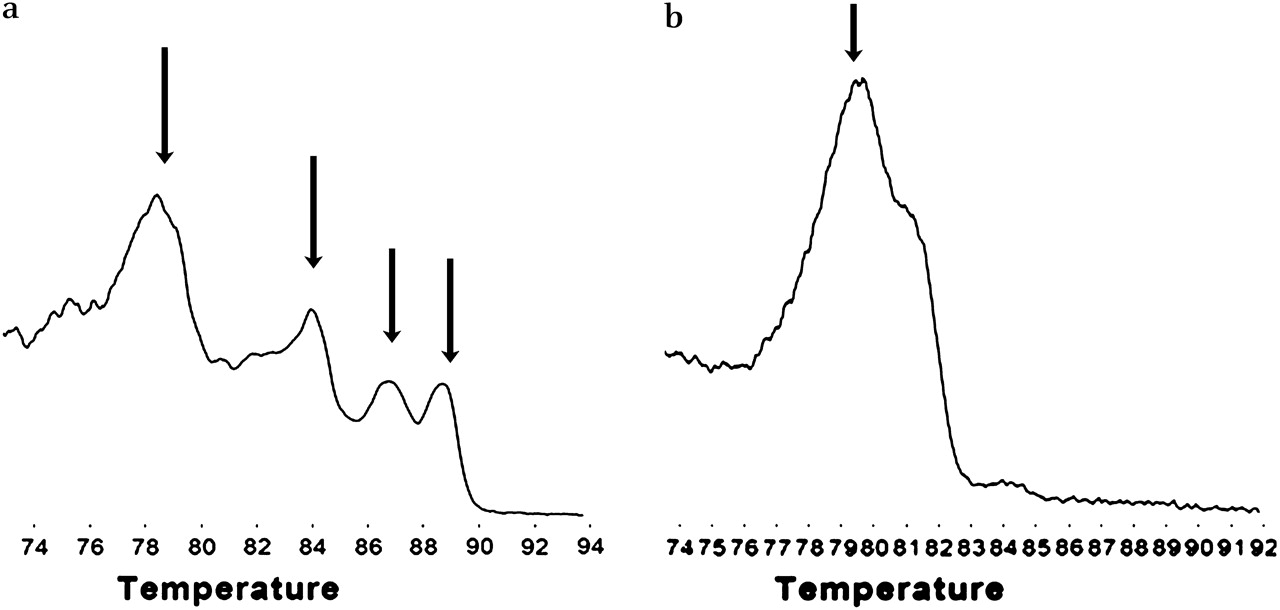
Template quality control consisted of multiplex PCR for four different sized fragments (100, 200, 300 and 400 bp) in the reference gene, GAPDH. As expected, when DNA extracted from cell lines was tested, all four bands were present. These are seen as specific peaks in the derivative plot produced by HRM analysis (figure 1). The FFPE samples produced variable numbers of bands reflecting variable DNA integrity. However, 7/50 (14%) of the samples (predominantly the older samples) did not show any amplification and were deemed unsuitable for PCR and not further analysed.
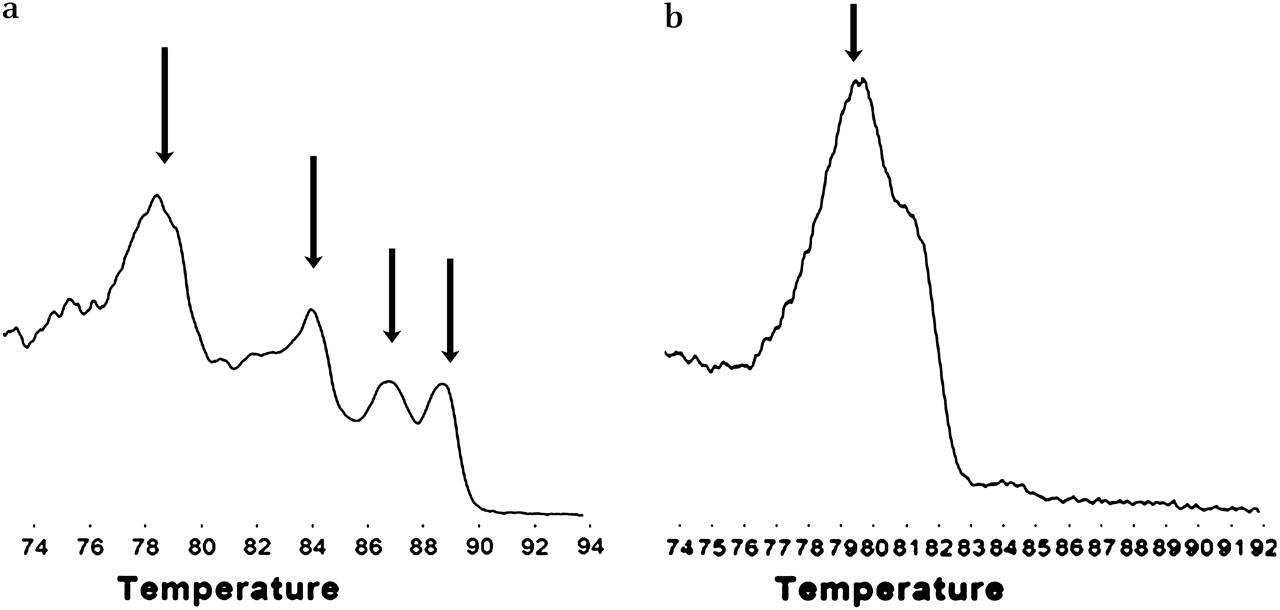
Testing the quality of DNA from formalin-fixed paraffin-embedded (FFPE) tissues. A multiplex PCR was performed, and the PCR products analysed using high-resolution melting after addition of LC green. (a) High-quality DNA from cell lines produced bands of 100, 200, 300 and 400 bp, which are seen as four peaks on the derivative plot (arrowed). DNA from the FFPE samples was of variable quality, and samples produced variable numbers of peaks. (b) Sample producing just a single peak of 100 bp. If there was no evidence of PCR amplification, the DNA was deemed too poor for PCR analysis.
QMC-PCR on DNA from cell lines
Mutation detection
Twenty-nine cell lines underwent direct sequencing analysis of the hotspots in KRAS and BRAF and were then screened by QMC-PCR. The results showed 100% sensitivity and specificity together with 100% positive predictive value (PPV) and negative predictive value (NPV) (figure 2a, tables 1 and 2). Cell lines were then screened for somatic mutations in 10 hotspots in KRAS, BRAF, PIK3CA and CDC4 using the QMC-PCR protocol and, of the 40 mutations detected, every single one was confirmed by sequencing (100% PPV). Thus, even though the PDM reaction contained 10 primer pairs, the QMC-PCR protocol with HRM showed outstanding performance when compared with direct sequencing.
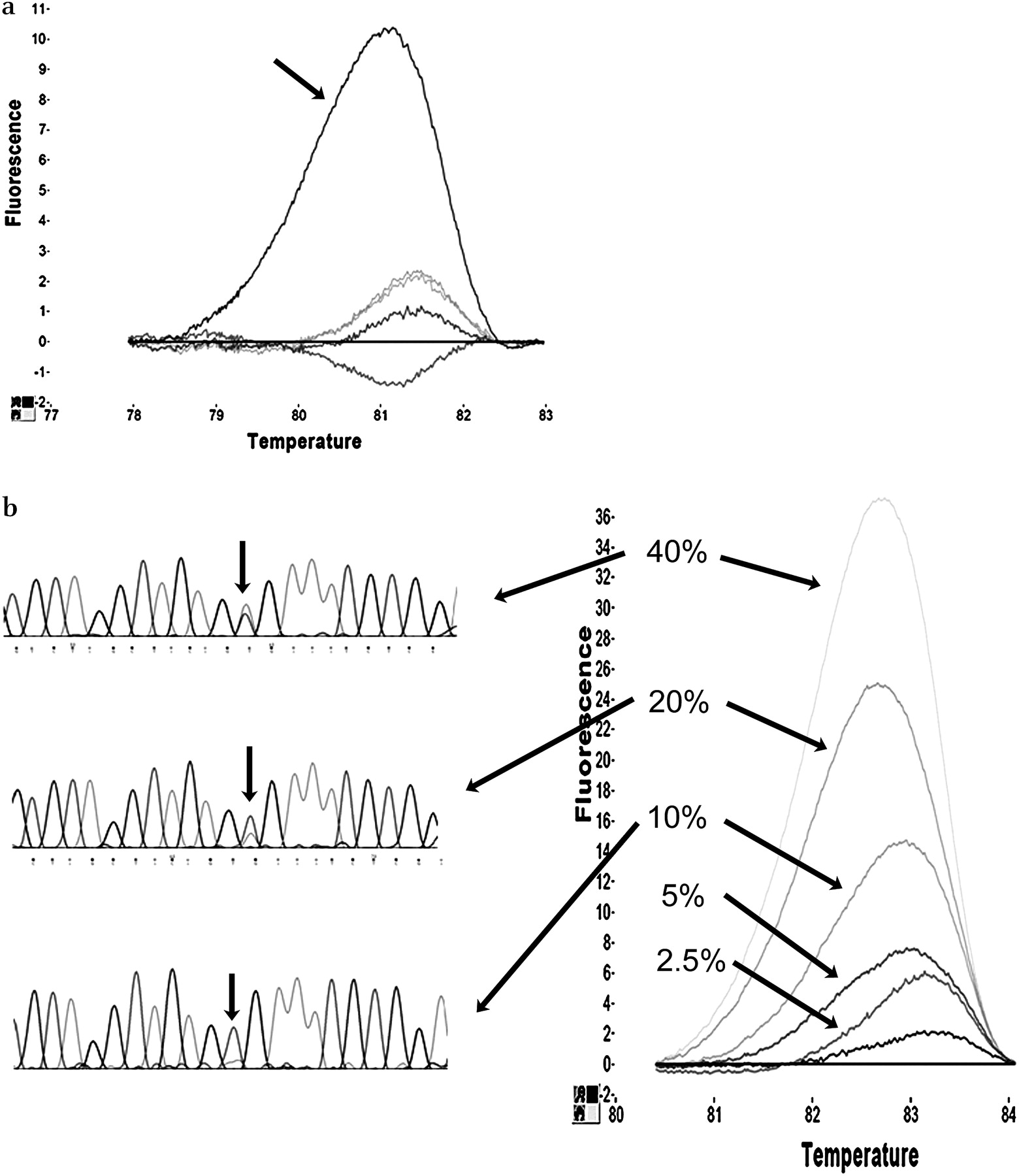
Optimisation and testing of quick-multiplex-consensus (QMC)-PCR. High-quality DNA from fingerprinted cell lines was used to optimise the QMC-PCR protocol. (a) Difference plot of BRAF exon 15 conducted on six cell lines showing how one of the cell lines containing a heterozygous mutation (HT29, arrowed) melts differently from the five wild-type alleles because of the formation of heteroduplexes. (b) Spiking experiments to test the limit of detection of QMC-PCR in comparison with direct sequencing. DNA from known mutant cell lines was spiked into DNA of known wild-types to produce mixtures containing 40%, 20%, 10%, 5%, 2.5% and 1.25% mutant alleles. The difference plot (for BRAF exon 15) shows that as few as 2.5% mutant alleles can be detected by HRM. In contrast, the chromatograms (on the left side) show that the mutant allele cannot be detected below the 20% level by direct sequencing.
Mutation detection. Quick-multiplex-consensus-PCR and high-resolution melting analysis
Mutation profile of cell lines
Limit of detection
To ascertain the limit of mutant allele detection, we performed a number of spiking experiments. DNA from DLD1 (containing an E545K mutation in PIK3CA) was spiked into DNA from VACO5 at different concentrations, and the resulting mixtures were screened for PIK3CA mutation using the QMC-PCR protocol. Similarly, DNA from VACO5 was spiked into DNA from HCT116, and the resulting mixtures were screened for BRAF V600E mutation. In both cases, mutations could be detected when as few as 2.5% mutant alleles were present (figure 2b). The spiked DNA mixtures were also tested by direct Sanger sequencing, and, in contrast with QMC-PCR, sequencing could not detect mutant alleles below a level of 20% (figure 2b). These data therefore show that QMC-PCR is nearly an order of magnitude more sensitive than direct sequencing for the detection of mutant alleles.
QMC-PCR on DNA from FFPE tissues
Comparison with standard PCR–HRM
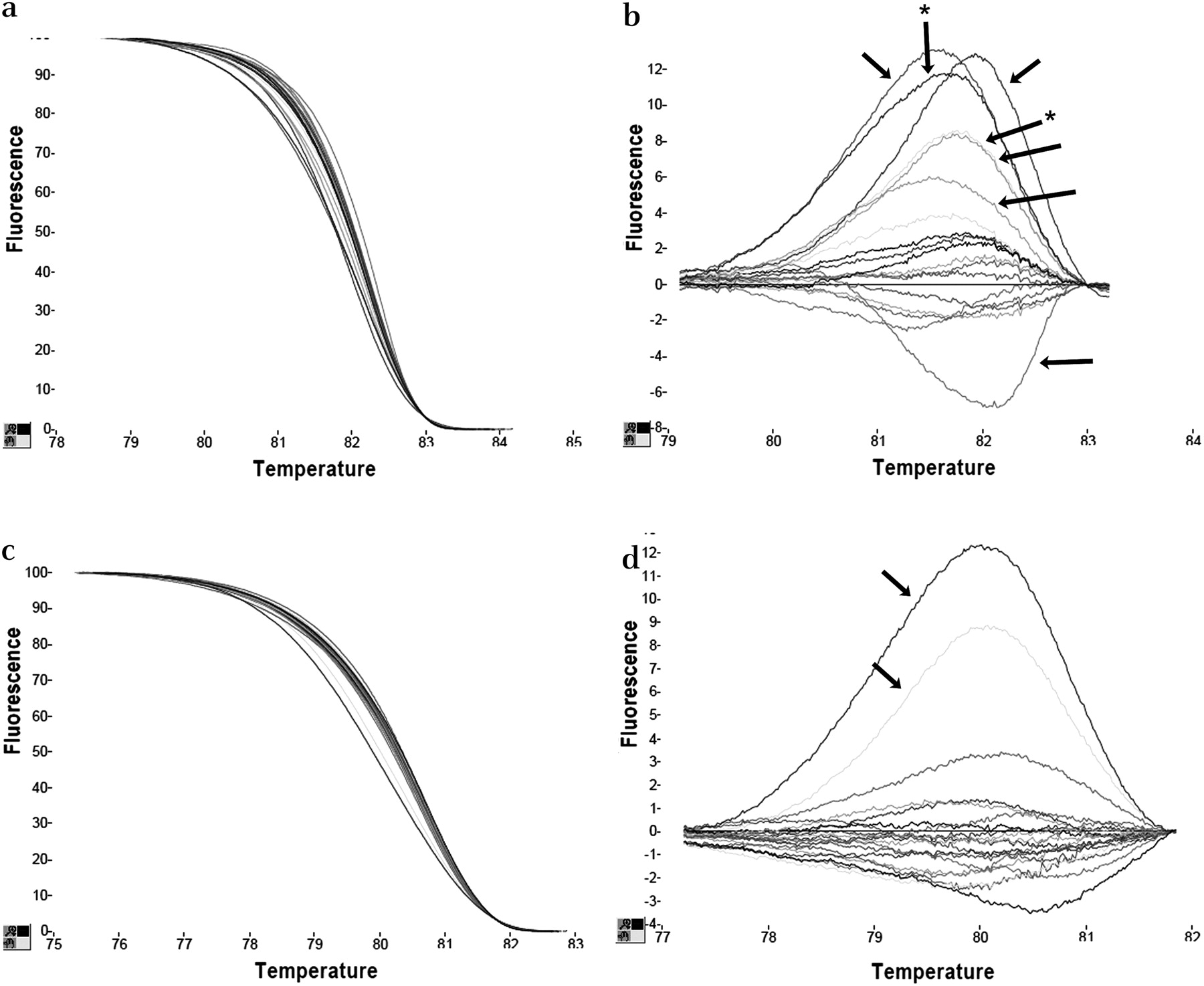
In order to be usable in the routine diagnostic setting, QMC-PCR must work with DNA derived from FFPE tissue. For any PCR-based technique, template quality is probably the most important factor, and thus we excluded seven low-quality DNA samples from further analysis. In the remaining 43 samples, a PCR product of at least 200 bp could be amplified. We firstly compared QMC-PCR and HRM with standard PCR and HRM because it is possible that QMC-PCR—especially with its multiplexed reaction—may result in poorer performance of the HRM technique. We tested samples for mutation in the BRAF exon 15 hotspot and found a much higher frequency of aberrant melting using the standard protocol than the QMC-PCR protocol. In a total of 43 tests, seven samples were positive by standard analysis (figure 3) but negative by QMC-PCR. However, the melting curve was often shifted to the right, indicating that melting was delayed—an unexpected finding as the melting of heteroduplexes should occur earlier and the curve should be shifted to the left. All samples were sequenced, and it was shown that the seven samples identified to be ‘mutant’ by standard analysis but identified as wild-type by QMC-PCR were in fact all wild-type. These data are consistent with those in the literature, which cites a false-positive rate of around 16% when standard PCR and HRM are used for mutation detection in FFPE tissue-derived DNA.15 These data also show that the QMC-PCR protocol eliminates the false-positives generated by standard techniques.

Quick-multiplex-consensus (QMC)-PCR eliminates false-positive artefacts generated with standard PCR when using DNA from formalin-fixed paraffin-embedded (FFPE) tissue. (a) shows that, with standard PCR, some samples show right-shifted melting curves and (b) shows marked changes on the difference plot suggesting, in this group of samples, that at least seven samples may contain mutations (arrowed). However, (c) shows that, with QMC-PCR on these samples, there were no right-shifted samples and (d) is the difference plot showing that only two were identified as mutant (denoted by * in (b)). Sequencing showed that QMC-PCR was correct.
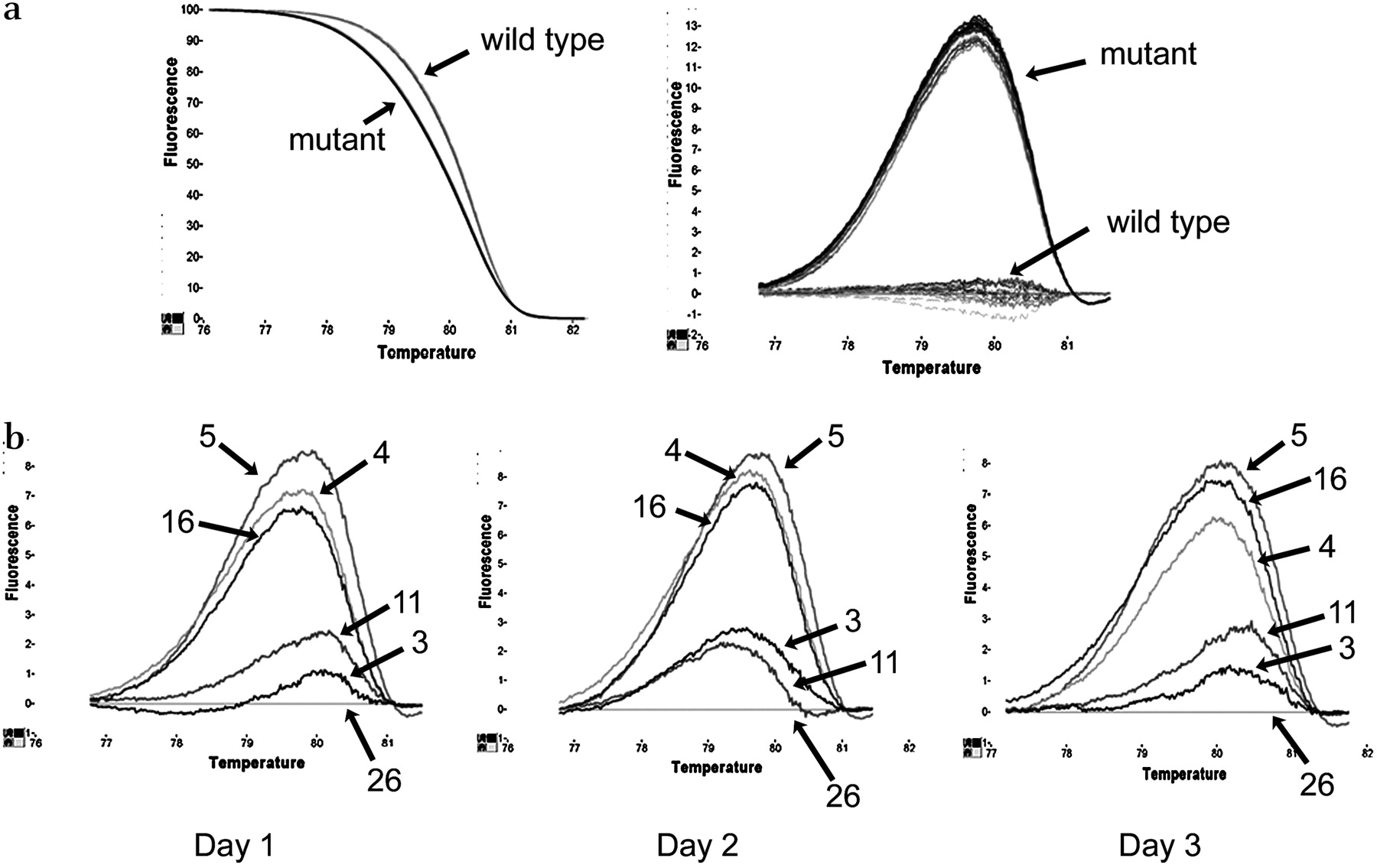
Assay precision
To test the robustness of the technique, we tested both long-term and short-term precision (intra-assay and interassay variation). For short-term precision, one hotspot in each of PIK3CA and BRAF was tested. Two cases—one mutant and one wild-type—were tested as 15 replicates and showed a very tight clustering together of the replicates (figure 4a). For long-term precision analysis, a series of (sequence-proven) wild-type and mutant samples for a hotspot in PIK3CA were tested on 3 consecutive days and yielded the same results on each occasion (figure 4b). These data show that this is a very robust and reproducible technique.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Testing reproducibility of quick-multiplex-consensus (QMC)-PCR. (a) Difference plot showing the intra-assay variation for PIK3CA exon 20. Two formalin-fixed paraffin-embedded (FFPE) samples (one mutant and one wild-type) were analysed as 15 replicates and showed negligible variation. (b) Interassay variation for the same target sequence using three mutant FFPE samples (4, 5 and 16) and three wild-types (3, 11 and 26) on 3 different days showing very little variation.
QMC-PCR as a screening tool
To test the utility of QMC-PCR with HRM as a screening test in DNA derived from FFPE tissue in comparison with direct sequencing, 43 samples underwent direct sequencing analysis for the hotspots in KRAS exon 2 and PIK3CA exon 20. In the former test, 15/43 (35%) were identified as mutant by QMC-PCR, whereas, in the latter test, 6/43 (14%) were noted as mutant (table 1). In these tests, QMC-PCR showed a sensitivity of 100%, specificity of 71%, PPV of 76% and NPV of 100%.
After this, all cases were screened for all 10 hotspots at least twice with no change in melting pattern on repeat, and, in 430 screening tests, a total of 43 (10%) potential mutations were identified. All cases considered mutant underwent direct sequencing, and 36/43 were thus confirmed, giving a PPV of 84%. However, the reproducible nature of the aberrant melting in the seven ‘false-positives’ and the fact that we have shown that the HRM technique is much more likely to pick up small numbers of mutant alleles than direct sequencing leads us to think that these cases probably represent sequencing ‘false-negatives’, with true mutations present in a minority population of cells.
Cold PCR on cell lines and FFPE tissue
The purpose of this study was to optimise protocols for mutation detection. Cold PCR has recently been described as a variation on the usual protocol that can enrich for mutant alleles. Although the cold protocols would significantly lengthen the mutation screening protocols, we reasoned that the time cost would be acceptable if it resulted in improved mutation detection. We empirically derived the Tc for PIK3CA exon 9 and exon 20 on our thermal cycler and tested both the full and the fast cold protocols on both spiked DNA mixtures and FFPE tissue-derived DNA. However, we found that cold PCR did not give any improvement in mutation detection over QMC-PCR (see supplementary data online).
Discussion
Demands for mutation detection in tumours will inevitably increase as more predictive mutations are identified. Tumour mutation profiling may become a routine part of the pathologist's workload, requiring technologies that work reliably on DNA derived from FFPE tissue. Furthermore, as the majority of cancer treatment is undertaken outwith research institutes, the methods must be implementable in district general hospitals. We have described QMC-PCR as a very rapid (about 100 min cycling time) and simple method which works robustly in FFPE tissue sections. It is a two-step nested procedure in which the first reaction is a PDM PCR containing up to 10 primer pairs. This is followed by a SSD reaction during which each of the hotspots are individually analysed. The fact that only a single protocol is used (ie, rapid cycling between 95°C and 55°C) means that multiple targets can be tested in a single run. If PCR machines that can capture fluorescence are used (such as certain real-time PCR machines), then multiple targets can be analysed simultaneously. The ease of data interpretation, the gel-free nature of the methodology, and the fact that the tests can be carried out on inexpensive equipment all represent significant advantages over more traditional methods of mutation screening.
We have shown that QMC-PCR works efficiently in both fresh and archival tissue. Using high-quality DNA from cell lines with known mutations in a variety of genes, we have shown that it has 100% sensitivity and 100% specificity. QMC-PCR has a limit of mutation detection that is nearly an order of magnitude better than the current ‘gold standard’ of Sanger sequencing. This more effective detection becomes important in cases in which a tumour shows heavy stromal infiltration which may ‘veil’ mutations with the less sensitive techniques. Furthermore, QMC-PCR compares very favourably with cold PCR because, in our hands, the cold protocols did not improve the level of detection. AS cold PCR requires that a Tc be optimised for each target and it is likely that the Tc may vary between machines, we consider our technique to be simpler.
In archival FFPE tissue, standard PCR followed by HRM is a technique prone to false-positive artefacts, thereby limiting its utility. QMC-PCR produced far fewer artefacts and led to a 16% increase in specificity. This is probably due to the nested nature of the procedure in which—in the SSD reaction—contaminants are diluted out and only specific PCR product is used as template. Compared with direct sequencing, we found that QMC-PCR has 100% sensitivity and did not miss any mutations. However, our study identified a number of clinical samples in which there was aberrant melting, but no mutations were detectable. These are theoretically regarded as false-positives, but, having demonstrated that QMC-PCR is more sensitive than direct sequencing, we think it likely that these cases contained small numbers of mutant alleles and that the sequencing data represent false-negatives. This does lead to a situation where the results of the screen cannot be validated by the current ‘gold standard’. It is possible that more sensitive sequencing techniques such as pyrosequencing may resolve this problem. On the basis of our experience, we would believe the QMC-PCR data over the sequencing data. Given the sensitivity of the technique, it may possible that HRM will become the new ‘gold standard’ for mutation screening.
We further tested the robustness of the method by evaluating variability both between and within assays. Tests performed on a number of DNA samples derived from FFPE tissue on 3 consecutive days gave identical results, showing that there was little interassay variation. Analysis of two samples (one wild-type and one mutant for PIK3CA), each tested as 15 replicates, resulted in near-identical melting curves of each of the replicates, thereby showing that the technique has high precision with negligible intra-assay variation.
Although QMC-PCR provides many advantages, there are some confounders in interpreting the data. Firstly, as the method was initially developed for SNP detection, these will be picked up when present as heterozygous variants and may be a source of false-positive results. Overinterpretation can be avoided through (1) analysis of the melting curves, as specific SNPs will produce specific melting patterns, and (2) running samples of non-tumour tissue from the same individual, which will show the presence of the SNPs. Ultimately, the aim of all screening methods is to identify those samples likely to be mutated for validation by sequencing. Thus sequencing samples with aberrant melting will ultimately discriminate SNPs from mutations.
In summary, we have described a new technique based on multiplexed nested PCR and HRM analysis. We have shown that the technique is rapid, robust and highly sensitive and, most importantly, it is easy to implement (requiring a single set of cycling conditions) and the data are easy to interpret. The technique allows multiple targets to be tested and it is ideal for tumour mutation profiling. Given that it does not need large sophisticated pieces of equipment, it can be established in almost any setting.
Take-home messages
High-resolution melting (HRM) is a cheap and reliable method for mutation detection.
Quick-multiplex-consensus (QMC)-PCR allows multiple mutation hotspots to be rapidly detected by HRM using a standardised nested protocol.
QMC-PCR has been developed specifically for formalin-fixed tissue and reduces the number of false-positive artefacts.
QMC-PCR with HRM is a more sensitive method for mutation detection than Sanger sequencing.
Acknowledgments
We thank Dr Simon Crook and Mr Darryl Jackson for assistance with cell culture, Dr Sally Chappell and Mr David Harris for initial help with HRM, and Annie Wilson and Carol Dunn for retrieving histopathology specimens. The study was supported by a grant from Nottingham University Hospital Charities.
References
Supplementary materials
Web Only Data jcp.2009.070508
Files in this Data Supplement:
Footnotes
Additional table and data are published online only at http://jcp.bmj.com/content/vol63/issue2
Funding University of Nottingham, Nottingham University Hospitals Charitable Trusts.
Competing interests None.
Ethics approval This study was conducted with the approval of the Oxfordshire REC B 05/Q1605/66.
Provenance and peer review Not commissioned; externally peer reviewed.