Article Text

Abstract
Aims—Comparative genomic hybridisation (CGH) is a reliable tool to gain an overview of all unbalanced chromosomal alterations within a tumour. Nevertheless, the high numbers of tumour cells required and the comparatively low resolution are drawbacks of this technique. Polymerase chain reaction (PCR) based multiplex microsatellite analysis represents a semi-automated, highly reproducible method, which requires small amounts of tumour cells. This is a comparative study of CGH and microsatellite analysis.
Methods—Eighty one samples of invasive breast cancer were investigated by two sensitive multiplex PCRs containing three microsatellites each of six markers (D6S261, D11S907, D6S300, D11S927, D8S272, and D11S925), and two additional microsatellite markers located within intron 1 of the epidermal growth factor recepter gene (egfr) and p53 (p53CA).
Results—At least one example of loss of heterozygosity was detectable in all breast cancer tissues. However, the overall rate of accordance between the two methods tested was only 61%. An increasing rate of the number of genetic alterations in each case was mirrored by a constantly increasing fractional allelic loss index.
Conclusions—PCR based multiplex microsatellite analysis using this panel of eight microsatellite markers not only enables the characterisation of cells that have malignant potential in a high frequency of patients with breast cancer, but can also give an estimate of the degree of genetic progression.
- breast cancer
- comparative genomic hybridisation
- microsatellites
- epidermal growth factor
- p53
Statistics from Altmetric.com
The origin of invasive breast cancer is still incompletely understood. Moreover, the disease displays a high degree of heterogeneity at the cytogenetic level, which can be detected by the method of comparative genomic hybridisation (CGH).1,2 CGH opened up the possibility of detecting all unbalanced cytogenetic alterations within tumour cells using a single approach. At the molecular level CGH can only detect chromosomal losses down to 8–10 Mb.3,4 Furthermore, the minimum amount of DNA, even in cases of microdissected tumours cells, is comparatively high and polymerase chain reaction (PCR) based DNA enrichment techniques need to be more reliable.5,6 The introduction of laser based microdissection techniques will mean that, in the future, research into breast cancer will concentrate increasingly on smaller cell populations; thus, methods with a low detection rate and a higher resolution will gain more importance. PCR based microsatellite analysis represents a semi-automated and highly reproducible method, which fulfills these requirements. Therefore, we applied a panel of highly informative microsatellite markers in a multiplex PCR approach, so that an estimate of genetic alterations in a few tumour cells could be made. Furthermore, this approach could provide evidence of small genetic alterations, which are not reflected at the cytogenetic level.7
To date, a direct comparison of genetic alterations revealed by CGH and multiplex microsatellite analysis covering multiple regions of the genome has not been performed. We aimed to investigate the relation between molecular–cytogenetic findings revealed by CGH and microsatellite analysis to evaluate the rate of reproducibility between those two methods and to develop a panel of microsatellite markers for multiplex PCR with high significance.
Material and methods
MATERIAL
Fresh frozen tissues from 81 invasive breast tumours were studied by microsatellite analysis. The tumours were staged as follows: 26 I, 34 II, and 21 III–IV, according to the American joint committee on cancer, 1993. Thirteen tumours were graded as grade 1, 37 as grade 2, and 31 as grade 3.
Tumour DNA was isolated with the QIAamp tissue kit from 10 sections with a thickness of 10 μm without prior microdissection. Leucocyte DNA used for the normal control was isolated with the QIAamp blood kit (Qiagen, Hilden, Germany) from EDTA blood from the respective patients. Tumour DNA from the same isolation step was used for CGH and microsatellite analysis.
CGH ANALYSIS
CGH analysis was performed in 31 tumours and the criteria for the evaluation of genetic alterations were as described previously.1,8,9 The tumours investigated by CGH were classified as ductal invasive G2 (n = 10), ductal invasive G3 (n = 11), lobular invasive (n = 3), tubular and tubulo-lobular (n = 2), and one case each of metaplastic invasive, tubular mixed, and mucinous invasive cancer.
MULTIPLEX PCR ASSAYS
Six microsatellite markers were grouped in two multiplex PCRs of three markers each. The DNA sequences of the microsatellites were obtained from the Genome Data Base, (www.gdb.org). Primers were created for multiplexing and synthesised by Biometra (Göttingen, Germany). Two multiplex microsatellite PCR protocols for a standardised assessment of loss of heterozygosity (LOH) in invasive breast cancer were developed. Six microsatellites were finally selected, using the following selection criteria: alteration frequencies of the informative cases in previous breast cancer studies of 10% and higher of all investigated cases (table 1), and a heterozygosity above 0.72. Both multiplex PCRs consist of three microsatellites with a difference in length of about 100 bp to prevent peak overlapping. The downstream primers of the two multiplex PCRs were labelled with different fluorescent probes for use in a single run analysis with a multicolour laser induced fluorescence capillary electrophoresis system. For standardisation of the analysis, the LOH score was calculated according to the equation published by Canzian et al.10 The reliability of peak area and lengths for LOH determination was tested by 10 runs of independent leucocyte PCR product showing a standard deviation (SD) of the quotient of the allele peak areas (a1/a2) of 7%. The critical difference was calculated by dc = 2 × √2 × SD,11 based on the previously determined SD. The cut off values for LOH were calculated by 1 − dc for the loss of the longer and by 1/ (1 − dc) for loss of the shorter allele, resulting in cut off values of 0.79 (loss of longer allele) and 1.27 (loss of shorter allele). Because of the low SD of the method, LOH could be detected sensitively if the peak area for one allele in relation to the other decreased in the tumour DNA to 79% of the normal. Therefore, a microsatellite was scored positive for LOH if R` was < 110% and the LOH score was > 1.27 or < 0.79. Multiplex PCR A consisted of the microsatellites D6S261, D11S907, and D6S300 using downstream 5`-FAM labelled primers and multiplex PCR B consisted of D11S927, D8S272, and D11S925 performed with downstream 5`-HEX labelled primers (table 1). Table 1 also shows the chromosomal localisation of the markers.
Loss of heterozygosity at eight polymorphic markers in 81 patients with invasive breast tumours
Multiplex PCRs were performed in a 100 μl reaction volume containing 30 pmol of each primer, 1 μl AmpliTaq GoldTM, 1 × GeneAmp™ buffer II, 2mM MgCl2, 200 μmol of each GeneAmp™ dNTP (Perkin Elmer, Weiterstadt, Germany), and 200 ng sample DNA. Investigations of the reaction conditions showed that the addition of 5% and 4% DMSO gave the highest product yield for multiplex PCR A and B, respectively. All PCR components except the sample DNA were premixed and applied to the patients' leucocyte and tumour DNA. Both multiplex microsatellite PCRs were performed with a Robocycler GradientTM (Stratagene, Uppsala, Sweden) under the same cycle conditions; namely: an initial seven minute denaturation at 95°C, followed by 42 cycles of one minute at 95°C (denaturation), one minute annealing at 57°C, and one minute extension at 72°C, with a final seven minute extension step at 72°C. Multiplex PCR A and B samples (2 μl) were simultaneously dissolved in 12 μl formamide with 0.5 μl standard, denatured for two minutes at 95°C, cooled down to 6°C for two minutes, and injected at 15 kV for five seconds. Separation was done with a four colour, laser induced fluorescence capillary electrophoresis system (Prism 310 Genetic Analyser), the polymer POP4, 1× genetic analyser buffer with EDTA (Perkin Elmer), and a 47 cm, 50 μm uncoated capillary (CS Chromatographie Service GmbH, Langerwehe, Germany) at 15 kV for 25 minutes. Evaluation was performed with the GenescanTM 2.1 evaluation software (Perkin Elmer).
P53CA PCR ASSAY
PCR of the p53CA microsatellite, which is directly located in the p53 gene,14 was performed in a 50 μl reaction volume containing 30 pmol of both primers, one labelled with 5`-FAM, 1× Gene Amp™ reaction buffer II, 1.5mM MgCl2, 200 μmol dNTPs, 0.3 μl Ampli Taq GoldTM (Perkin Elmer), and 200 ng sample DNA. All components except DNA were premixed and added to the tumour and to the leucocyte DNA of the patients. Cycling conditions were as follows: initial denaturation at 95°C for nine minutes, followed by 36 cycles of one minute at 95°C, one minute annealing at 56°C, and one minute extension at 72°C, and a final seven minute extension step at 72°C. PCR products of leucocytes and tumour for each patient were tandemly separated with the Prism 310 genetic analyser (Perkin Elmer) using Genescan-500TM TAMRA 500 length standard, POP4 (Perkin Elmer), a 47 cm uncoated capillary (CS Chromatographie Service), and 15 kV for 18 minutes.
INTRON 1 EGFR ALTERATIONS
Alterations within a CA repeat in intron 1 of the epidermal growth factor receptor (egfr) gene were determined as described previously.7
STATISTICAL TESTS
Statistics were performed using the Kruskal-Wallace test.
Results
ALTERATION FREQUENCIES DETERMINED BY MICROSATELLITE PCR
For standardisation of the analysis, LOH scores were calculated according to the equation published by Canzian et al (see methods section).10
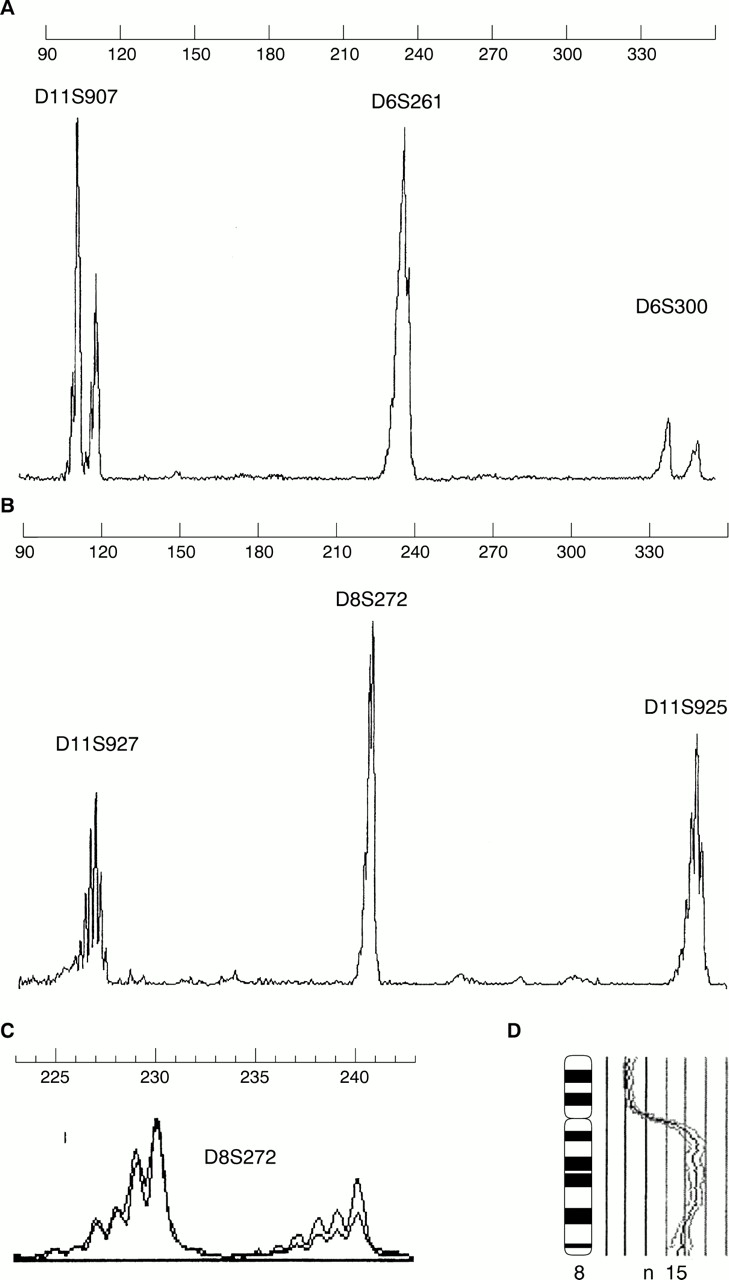
Using these methods, we investigated invasive breast tumours from 81 patients by multiplex PCR for eight polymorphic microsatellite markers (table 1; fig 1) and at least one example of LOH detected in all tumours. D11S925 (50%) and D8S272 (62%) showed the highest alteration frequencies, followed by D6S261 (46%), D6S300 (41%), D11S927 (38%), and D11S907 (35%). LOH of p53CA was detected in 54% and within the CA repeat in intron 1 of egfr in 39% of all informative cases.

(A, B) Two multiplex microsatellite PCR runs. (C) Loss of heterozygosity of the second allele of D8S272 (score 0.52). Comparative genomic hybridisation showing amplification at chromosome 8q.
ALTERATION FREQUENCIES DETERMINED BY CGH
Tumours of 31 patients were investigated by CGH. Losses of 8q were not detected, in contrast to 8q gains (38%). Loss of chromosomal material at 11q was seen in 38%, at 6q in 16%, at 7p in 9%, and at 17p in 19% of the cases. Table 2 gives an overview of all chromosomal changes detected with a frequency of at least 15%, demonstrating the representivity of the investigated tumour series.
Chromosomal losses and gains with a frequency of at least 15%
COMPARISON OF MICROSATELLITE PCR AND CGH
All eight markers showed an overall rate of 61% accordance with CGH results. The highest rates of accordance were observed in D6S300 (72%), p53CA (72%), D11S925 (68%), and D11S927 (60%), followed by D11S907 (60%), D6S261 (58%), egfr intron 1 CA repeat (47%), and finally D8S272 (41%). Remarkably, 35% of the cases with LOH of D8S272 revealed an amplification or high level gains, including 8q23–24 (fig 1 C, D). Assuming that LOH in a tumour is mirrored by a loss of the respective chromosomal arm, CGH analysis ascertained this result in 0% (D8S272, D11S907), 9% (intron 1 CA repeat of egfr), 50% (D11S927, D6S261), 57% (D6S300, p53), and 69% (D11S925) of cases.
An increasing, but not significant (p < 0.2), rate of fractional allelic loss, defined as numbers of markers with allelic loss divided by the numbers of markers that were informative,12 was related to a constantly increasing average number of cytogenetic alterations in each case measured by CGH (fig 2).

{kind=link}
{kind=link}
Average number of genetic alterations in each case plotted against the fractional allelic loss. As the fractional allelic loss increases, the number of genetic alterations increases also.
Discussion
Invasive breast cancer has been investigated extensively using a variety of cytogenetic and molecular genetic approaches, such as CGH, fluorescence in situ hybridisation (FISH), and PCR based microsatellite analysis. Unfortunately, the comparability of the methods is still poor. For instance, one previous study showed that conventional metaphase cytogenetics results can be reproduced in only 50–60% of the identical tumours using CGH.13 Furthermore, high level gains and amplifications, as detected by CGH, revealed misinterpreted LOH results obtained by microsatellite analysis.2,14
Microsatellite and CGH analyses are two methods with different resolutions that are used to evaluate the genotype of cancer cells. The question could be raised as to whether alterations detected by these methods are features of a chromosomal instability phenotype,12 which might be caused by similar mechanisms. Chromosomal instability is a feature of invasive breast cancer and it is noteworthy that our panel of eight highly informative microsatellite markers was adequate to detect at least one LOH in all the breast cancer cases. Interestingly, the numbers of alterations in each case increased in parallel with the fractional allelic loss index (fig 2). Therefore, this panel is not only a valuable tool for detecting LOH with a sufficient success rate, but also gives an approximate impression of the degree of chromosomal instability within a distinct tumour.
Regarding the above mentioned data only, one would expect the degree of accordance between the two methods to be reasonably high. Surprisingly, the results obtained by CGH could be reproduced by microsatellite analysis in only 61% of the cases. It has to be stressed that the genetic alterations of interest in the tumours analysed by CGH were detected in a similar ratio as seen in earlier published studies,2,9 and therefore the cohort of tumours analysed by CGH is assumed to be representative.
The highest degree of discrepancy was observed in D8S272. High level gains and amplifications of chromosomal material of 8q, including 8q22–24, are some of the most frequent cytogenetic alterations in breast cancer, and might be misinterpreted as LOH in D8S272 microsatellite analysis. This mechanism may also be responsible for the discrepancies at the 11q13, 17q21–24, and 6q loci, which are as important in invasive as in in situ breast cancer.1,2,14–17 Therefore, in cases with LOH or allelic imbalances (which is a better term for this biological phenomenon), the results need to be confirmed using additional techniques such as FISH or real time PCR. Real time PCR has the advantage that it can be performed using small numbers of tumour cells and the need for reference DNA can be circumvented. In addition, this method will give exact information about the gene dosage of a given gene.
Another discrepancy appearing at a higher frequency was the detection of LOH by microsatellite analysis but no detectable chromosomal loss by CGH. The percentages of detection failure by CGH within distinct chromosomal regions varied greatly from 30% (D11S925) to 90% (CA repeat in intron 1 of egfr). One possibility could be the different thresholds used for the definition of a loss of genetic material. Taking into account the cut off values for LOH detection by microsatellite analysis and the CGH threshold for the definition of loss of genetic material in our study, microsatellite analysis might be over sensitive for the detection of genetic losses. However, the re-evaluation of the respective CGH ratio profiles with weaker threshold limits did not suggest that this could be a sufficient explanation. It has been shown that deletions in egfr in invasive breast cancer are not reflected by cytogenetic changes.7 A similar feature is observed in colon cancer with regard to 3p21–22—the chromosomal locus of transforming growth factor receptor β2 (TGFR-β2).18,19
Further detailed analysis shows that only a minority of tumours with discrepant results show a loss of genetic material by CGH and no LOH by microsatellite analysis in a distinct chromosomal region. All these cases revealed an aneuploid DNA pattern (data not shown), which probably characterises these genetic events as secondary as a result of genetic progression, with both original alleles unaffected.
Therefore, these results point to the power of microsatellite analysis in the detection of additional, submicroscopical genetic alterations, even in 150–200 tumour cells.20 An additional advantage of this method over CGH is that it allows the investigation of highly fragmented DNA (DNA fragments of ≤ 1000 bp), often obtained from formalin fixed, paraffin wax embedded tissue. Because this technique does not merely reflect gross cytogenetic changes, it appears to be one of the methods of choice for the investigation of small numbers of tumour cells.
Acknowledgments
We thank U Neubert, P Meier, L Grote, A Stefanowicz, and W Bücker for technical assistance in CGH analysis and multiplex PCR analysis. Grants: Deutsche Krebshilfe, 10–1681-Bü-I; University of Münster (IMF), Bü 1 1 98 31.