Article Text

Abstract
A 67 year old man presented with a polypoid lesion on the temple that had all the light microscopic, immunohistochemical, and ultrastructural features of a rhabdoid tumour. There was an area of intraepidermal carcinoma and invasive squamous carcinoma at the base of the polyp. The tumour progressed aggressively and the patient died five months after primary excision. Cutaneous tumours with a rhabdoid morphology have been described previously and tend to have a very poor prognosis. No previously published report describes a clear squamous histogenesis.
- rhabdoid
- skin
- squamous carcinoma
Statistics from Altmetric.com
Malignant rhabdoid tumours were originally described in the kidneys of children in 1978, as a subset of the first national Wilms's tumour study.1 Since that time, identical tumours have been described in a variety of extrarenal organs and soft tissues with a broader range of patient ages. Recently, “composite” extrarenal malignant rhabdoid tumours—in which a recognisable parent neoplasm is admixed with a malignant rhabdoid tumour—have been observed.2,3 Such tumours suggest that the rhabdoid morphology might be a common endpoint in the clonal evolution of tumours of different origins. The recognition of the rhabdoid morphology in a pure or composite tumour is important, because it appears to be associated with aggressive biological behaviour, rapid dissemination, and poor response to adjuvant treatment.2–4
Malignant rhabdoid tumours of the skin have been described only rarely. Most occur in children and are associated with a rapidly fatal outcome. Malignant melanomas with a predominant rhabdoid morphology have been seen in adults. Our case represents a composite malignant rhabdoid tumour that appears to have evolved on a background of intraepidermal carcinoma and invasive spindle cell squamous carcinoma of the skin. We believe this to be the first such case reported in the English language literature.
Case history
A 67 year old white man presented in May 1998 with a rapidly growing polypoid lesion on the right temple, which obstructed the lateral visual field. This was excised as an emergency because of superinfection and bleeding. The lesion had evolved from a longstanding keratotic patch, and had grown over a period of three to four months. There was no evidence of local lymphadenopathy at the time of presentation.
Past medical history included chronic renal failure secondary to microscopic systemic vasculitis, and type II diabetes mellitus. Drug treatment included azathioprine and long term oral prednisolone.
Macroscopic findings
The biopsy specimen consisted of a superficially ulcerated pedunculated lesion measuring 6.5 × 6.0 × 2.5 cm, with a small stalk covered by residual skin at one pole (fig 1). On cross section the tumour had a uniform cream/white fish flesh appearance. Representative tissue portions were fixed in 10% neutral buffered formalin, routinely processed, and embedded in paraffin wax. Tissue sections were stained with haematoxylin and eosin.
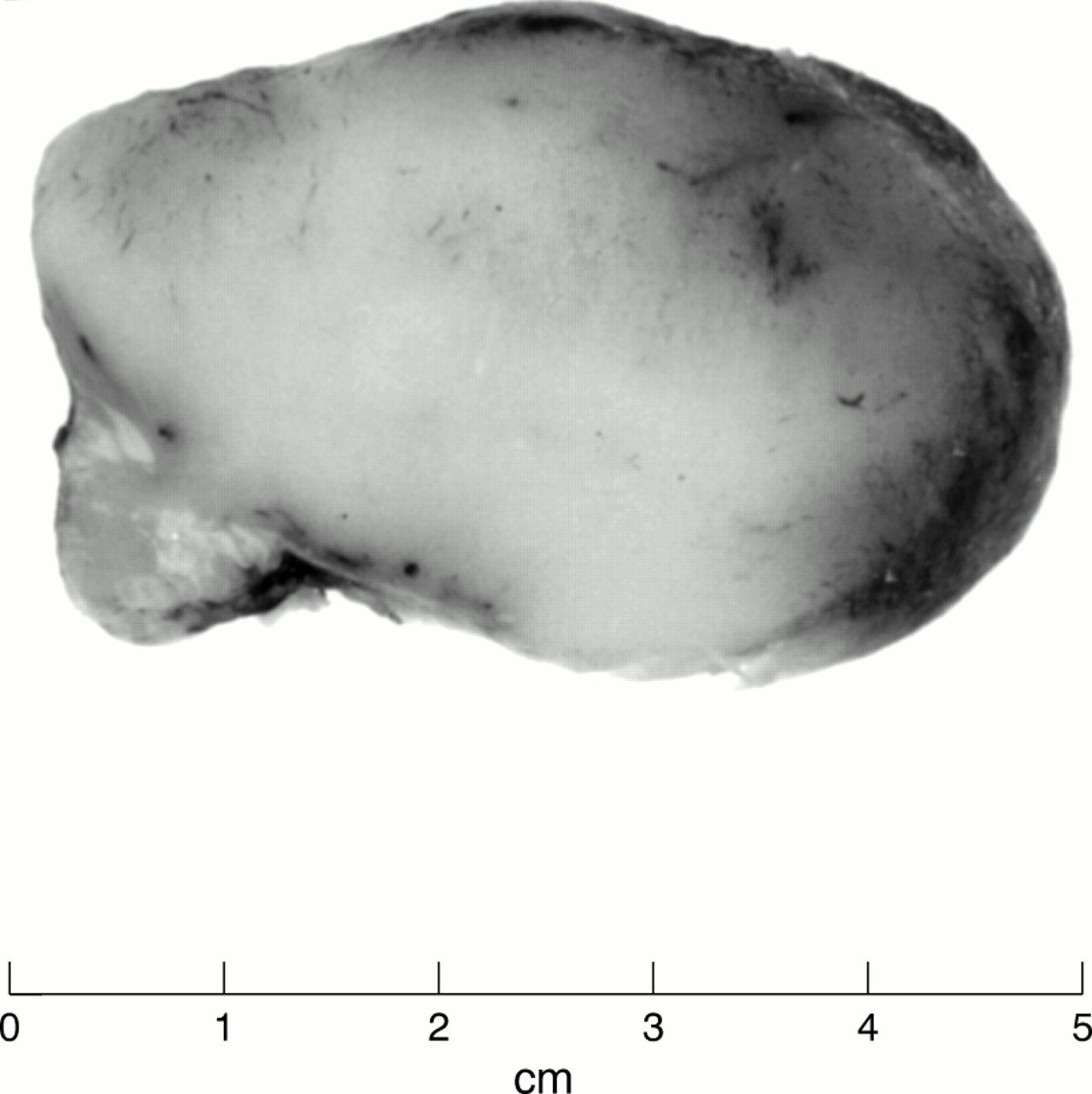
The macroscopic appearance of the biopsy specimen showed a pedunculated tumour with uniform white cut surfaces.
Light microscopic findings
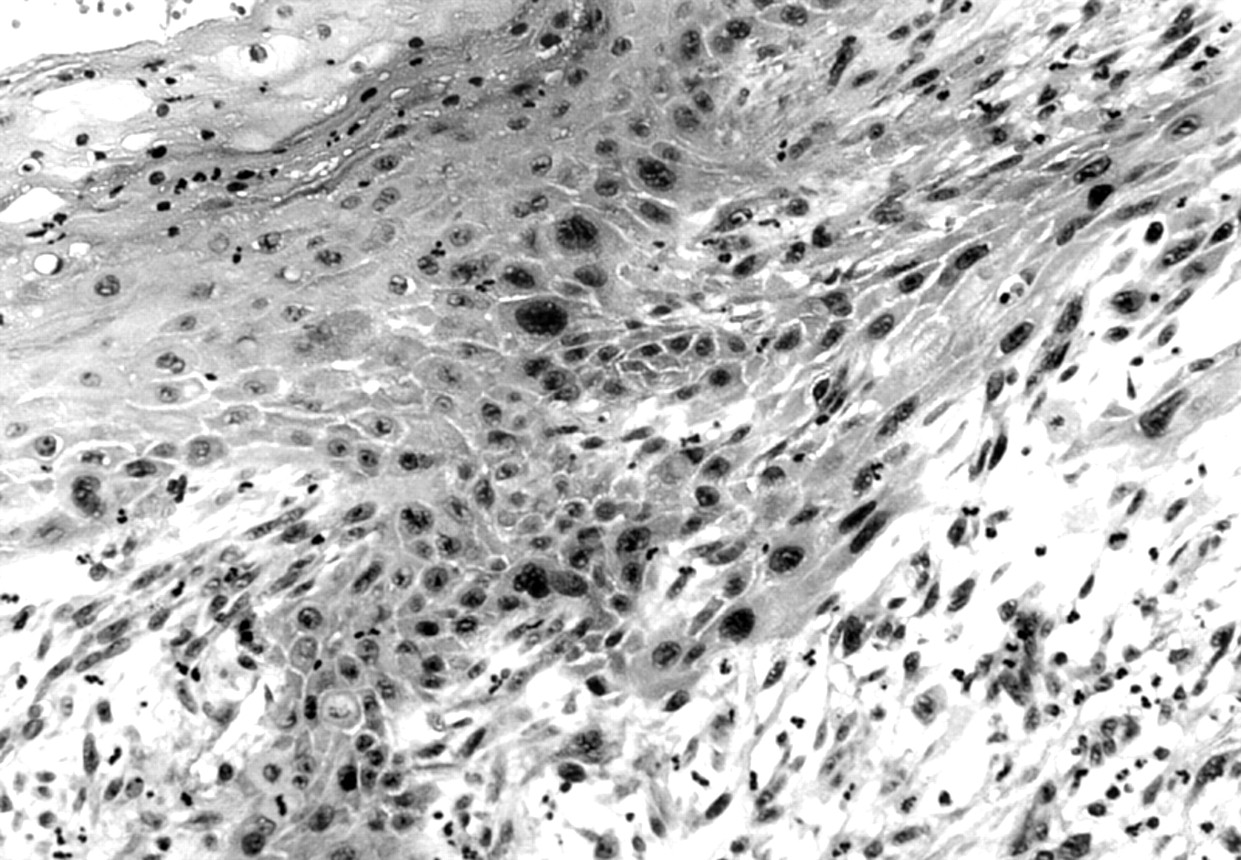
Histological examination of the small stalk with intact skin at the base of the lesion showed evidence of intraepidermal carcinoma, and a focus of poorly differentiated spindle cell squamous carcinoma (fig 2). However, the polypoid portion of the lesion had a very different appearance. This was composed of monomorphic polygonal cells arranged both in solid sheets and in an alveolar pattern with inconspicuous stroma. The cells had large vesicular nuclei with prominent nucleoli. In addition, there were prominent homogenous eosinophilic cytoplasmic inclusions, which caused lateral displacement of nuclei, giving a characteristic rhabdoid appearance (fig 3).

Microscopy showing an area of intraepidermal carcinoma and an invasive component present at the base of the pedunculated neoplasm.
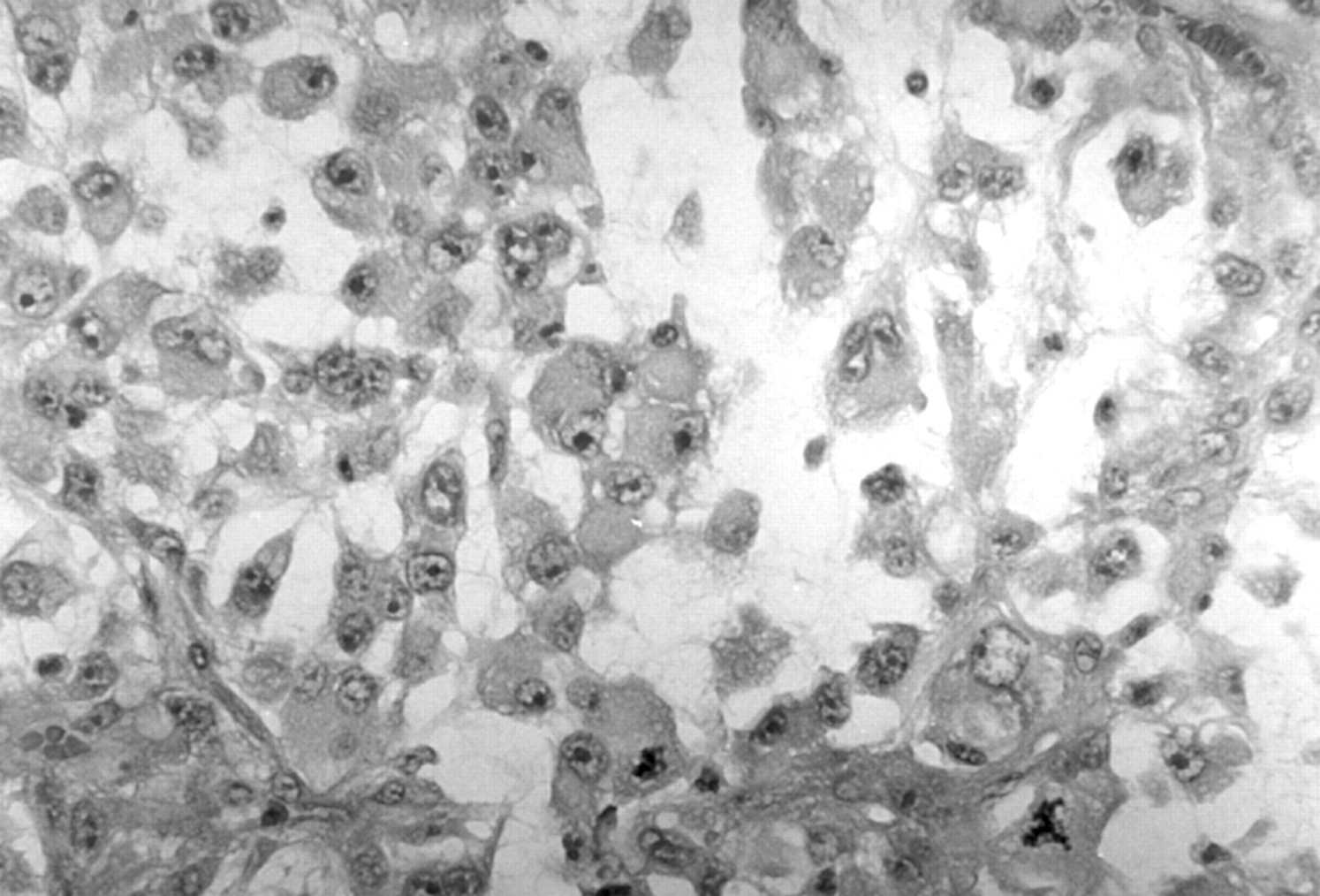
Representative section of the skin tumour showing discohesive rhabdoid cells with characteristic eosinophilic inclusions and large eccentrically placed nuclei (haematoxylin and eosin stained).
Immunohistochemical findings
Sections were stained with antibodies to cytokeratin, vimentin, epithelial membrane antigen (EMA), and S100.
There was strong coexpression of cytokeratin and vimentin, localised to the cytoplasmic inclusions within the tumour cells. There was no expression of EMA or S100.
Electron microscopy
Tissue from the tumour was processed for electron microscopic examination. The most striking feature was that of focal accumulations of intermediate filaments within the cytoplasm, corresponding to the eosinophilic inclusions seen on light microscopy (fig 4). In addition, there were primitive attempts at desmosome formation between contiguous tumour cells, and a moderate amount of dilated rough endoplasmic reticulum was seen.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Electron micrograph showing characteristic accumulation of intermediate filaments in the cytoplasm of cells with a rhabdoid morphology.
Clinical follow up
One month after the primary excision a further wide local excision with rhomboid flap was performed for residual disease, leaving a 10 mm margin of clearance. Histological examination of this specimen showed similar features to the first biopsy.
At review one month later, four separate tumour nodules were identified, three at the excision margin and one on the skin of the upper cheek. There was pronounced swelling in the preauricular and upper cervical areas. A computed tomography scan the next week showed recurrent disease, with satellite nodules around the area of excision, and a lymph node mass extending from the parotid to the upper neck.
The patient received a course of palliative radiotherapy but within a month the tumour had again grown rapidly around the orbit and in the preauricular area. He was discharged to hospice care and died five months after the primary excision.
Discussion
Beckwith and Palmer first identified tumours with a rhabdoid morphology within the kidneys of children in 1978.1 Clinically, their delineation from other paediatric renal neoplasms is important because they are associated with a poor prognosis. Subsequently, phenotypically identical tumours have been described at a variety of anatomical sites. These tumours occur most often in children and are associated with a similarly rapid fatal outcome.
Malignant tumours with a rhabdoid morphology rarely occur in adults and many of those described appear to represent clonal evolution in malignant tumours of varying histogenesis. In these cases, areas of the primary tumour or metastatic deposits have a rhabdoid morphology, whereas other areas have the morphology of the parent neoplasm, such as carcinoma, melanoma, or sarcoma.2,3 Recognition of the rhabdoid morphology in these tumours is important clinically, because there is evidence that they have the same aggressive biological behaviour as those tumours seen in children. Furthermore, they do not appear to respond to any currently available adjuvant treatment.1,2
Malignant rhabdoid tumours of cutaneous origin are very rare. The first report of primary presentation in the skin was by Dabbs and Parks in 1988.5 Interestingly, this was also a case of a rapidly growing lesion on the temple of an elderly man, with early involvement of local lymph nodes. Most cases in the literature have occurred in young children and were associated with a rapidly fatal outcome.6 Some of the cases described in adults were malignant melanomas with a rhabdoid morphology,7,8 but in others the histogenesis was uncertain.9 Our case had a classic rhabdoid morphology but the base of the tumour showed a focus of invasive spindle cell squamous carcinoma, whereas the adjacent epithelium showed the changes of intraepidermal carcinoma. We believe this to be the first case of cutaneous malignant rhabdoid tumour showing clear squamous histogenesis.
Anecdotally from colleagues, and from personal observation, we have noted an association between the development of similar appearing tumours at other sites and a history of long term immunosuppressive treatment, as in this case. It is possible that this might play a role in the development of these lesions and this could benefit from further investigation.