Article Text

Abstract
Background: The release of matrix degrading enzymes such as matrix metalloproteinase 9 (MMP-9) from bronchial epithelial cells is critically involved in airway wall remodelling in chronic inflammatory processes of the respiratory system. MMP-9 expression is induced by inflammatory mediators such as tumour necrosis factor (TNF)-α, but to date nothing is known about the mechanisms of inhibition of MMP-9 expression in these cells.
Methods: A study was undertaken to examine whether activators of the nuclear transcription factor peroxisome proliferator activated receptor gamma (PPARγ) might modulate MMP-9 expression in two different bronchial epithelial cell lines.
Results: PPARγ was expressed and was functionally active in NL20 and BEAS cells. Activation of PPARγ by rosiglitazone or pioglitazone significantly reduced TNF-α and PMA induced MMP-9 gelatinolytic activity in a concentration dependent manner in both cell lines, but did not alter the expression of tissue inhibitor of MMPs type 1 (TIMP-1), the local inhibitor of MMP-9. Northern blot analysis revealed a decrease in MMP-9 mRNA expression following treatment with PPARγ which resulted from the inhibition of NF-κB activation in these cells, as determined by transient transfection assays and electromobility shift assays.
Conclusion: Activation of PPARγ in human bronchial epithelial cells limits the expression of matrix degrading MMP-9. This might have therapeutic applications in chronic inflammatory processes of the respiratory system.
- matrix metalloproteinase (MMP)-9
- peroxisome proliferator activated receptor (PPAR)γ
Statistics from Altmetric.com
Chronic inflammatory processes in the respiratory system such as bronchial asthma are characterised by airway wall remodelling with degradation and synthesis of interstitial matrix proteins and migration of bronchial epithelial cells.1,2 Degradation of collagen type IV is a critical step in the inflammatory disorganisation of the airway wall, and is mainly determined by the balance between matrix degrading matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of MMPs (TIMPs). Among them, gelatinase A (MMP-2) and gelatinase B (MMP-9), as well as TIMP-1, released from bronchial epithelial cells seem to be crucially involved in the pathogenesis of bronchial asthma.3 MMP-9 is increased in bronchoalveolar lavage fluid4 and bronchial tissue in patients with bronchial asthma,5,6 while the expression of TIMP-1 is increased to a lesser extent, thus shifting the balance towards matrix degradation in the airway wall. Various proinflammatory mediators such as tumour necrosis factor (TNF) and platelet activating factor (PAF) released from inflammatory cells in the airway system are potent inducers of MMP-9 but not MMP-2 expression.7 Recent work has shown that TNF-α induced MMP-9 expression in bronchial epithelial cells is regulated by activation of the proinflammatory transcription factor NF-κB.7 Since matrix degradation by MMPs is considered to be a major contributor to airway remodelling in bronchial asthma, the inhibition of MMP expression might preserve the histological airway structure in chronic inflammatory processes. To date, however, little is known about such counterbalancing mechanisms.
Peroxisome proliferator activated receptor gamma (PPARγ) is a nuclear transcription factor originally described as a major regulator in glucose homeostasis and adipogenesis,8 but recent work has shown that PPARγ activation might exhibit anti-inflammatory properties in different chronic inflammatory processes.9,10 PPARγ belongs to the group of nuclear hormone receptors consisting of a ligand and DNA binding domain which, upon activation by their respective ligands, bind to specific PPAR response elements (PPREs) in the promoter of their target genes, thus regulating their expression.11 PPARγ can be activated by naturally occurring ligands such as the prostaglandin D2 derivative 15-deoxyΔ12,14PGJ2,12 as well as by a group of new antidiabetic agents—thiazolidinediones (TZDs or glitazones) such as rosiglitazone or pioglitazone.13 PPARγ induces the expression of genes such as lipoprotein lipase and Glut4 and has been shown to limit the expression of proinflammatory mediators such as cytokines and chemokines. Some of these effects were mediated by an interaction with proinflammatory transcription factors like AP-1 or NF-κB.14,15 In addition, PPARγ has been shown to inhibit the expression and activity of MMP-9 in monocyte/macrophages and vascular smooth muscle cells through an as yet undefined mechanism.16,17 Previous work has demonstrated PPARγ expression in bronchial epithelial cells in situ and suggested an antiproliferative effect in these cells.18,19 However, to date nothing is known about the role of PPARγ in the regulation of MMP expression in bronchial epithelial cells.
We hypothesised that PPARγ is functionally active in human bronchial epithelial cells and that PPARγ activators might modulate the expression of MMP-9 in these cells.
METHODS
Cell culture
Normal human bronchial epithelial cells (cell line NL20) were obtained from ATCC and the human bronchial epithelial cell line BEAS-2B was provided by Berthold Fischer, Department of Internal Medicine II, University of Mainz. Both cells lines were established by transfection with the origin of replication defective SV40 large T plasmid. NL20 cells were cultured in Ham’s F12 medium with supplements according to the manufacturer’s protocol. BEAS-2B cells were cultured inRPMI supplemented with penicillin, streptomycin, and 10% fetal calf serum. Primary human bronchial epithelial cells were obtained from Biowhittaker and cultured according to the manufacturer’s protocol.
RT-PCR
Total RNA from NL20 and BEAS cells was isolated for RT-PCR with amplification of PPARγ and GAPDH cDNA as described previously.17
Gelatin zymography and ELISA
To examine the effect of PPARγ activators on MMP-9 gelatinolytic activity, cells were treated with TNF-α (10 ng/ml) or PMA (10 ng/ml) in the absence or presence of two different PPARγ activators—rosiglitazone (GlaxoSmithKline) and pioglitazone (Takeda)—in serum-free media for 12 hours. Culture supernatants were mixed in SDS-PAGE loading buffer (lacking reducing agents), applied to 10% SDS-polyacrylamide gels containing 1 mg/ml gelatin (Bio-Rad), and separated by electrophoresis. Subsequently, SDS was removed from the gels by two washes (15 minutes) with 2.5% Triton X-100 (VWR Scientific). After the washes, gels were incubated overnight (37°C) in zymography buffer (50 mmol/l Tris, pH 7.3, 10 mmol/l CaCl2, and 0.05% Brij 35 (Sigma)) and stained with Coomassie brilliant blue (Sigma). Gelatinolytic activity was visualised as clear zones of lysis against a dark background. In some experiments cells were treated with TNF-α and PPARγ activators for 12 hours, then cultured again in regular media for 12 hours before additional stimulation with TNF-α in the absence or presence of rosiglitazone or pioglitazone.
TIMP-1 protein content in conditioned media was determined by ELISA according to the manufacturer’s instructions (R&D Systems).
Northern blot analysis
Total RNA (5 μg) was used for standard northern blotting.
Transient transfection assays
To examine whether PPARγ is functionally active in the cells used, NL20 and BEAS cells were transiently transfected with PPRE3-TK-LUC20 (provided by Dr Bruce Spiegelman, DFCI, Boston, MA, USA) and pCMV-β-GAL using superfect, according to the manufacturer’s protocol (Quiagen). Transfected cells were stimulated with rosiglitazone at the indicated concentrations. Cells were harvested after 24 hours and luciferase and β-galactosidase activity was measured using the Dual-Light assay (Tropix, Bedford, MA, USA).
To investigate the effect of PPARγ activators on NF-κB activity, BEAS cells were transiently transfected with a luciferase reporter construct containing three copies of the prototypic NF-κB site of the MHC I promoter and a pCMV.β-GAL construct as an internal control. Transfected cells were cultured for 16 hours before stimulation with TNF-α, with or without PPARγ activators. Cells were then harvested after 6 hours and lysates were subjected to luciferase and β-galactosidase assay (Tropix).
Electrophoretic mobility shift assay
For electrophoretic mobility shift assays (EMSA), human BEAS cells were stimulated for 2 hours with TNF-α (10 ng/ml) in the absence or presence of PPARγ activators before nuclear extracts were prepared. Standard EMSAs were performed, using an oligonucleotide (Genosys Biotechnologies, The Woodlands, TX, USA) spanning a prototypic κB site from the MHCI promoter.
Statistical analysis
The results of the experimental studies are reported as mean (SE). Differences were analysed by one way ANOVA followed by the appropriate post hoc test. A p value of <0.05 was regarded as significant.
RESULTS
Expression of PPARγ in human bronchial epithelial cells
RT-PCR was performed to establish the expression of PPARγ in human bronchial epithelial cells used in our experiments. NL20 cells and BEAS cells contained PPARγ mRNA as detected by a 376 bp RT-PCR product (fig 1A). The identity of the detected PPARγ bands was confirmed by DNA sequencing (data not shown).
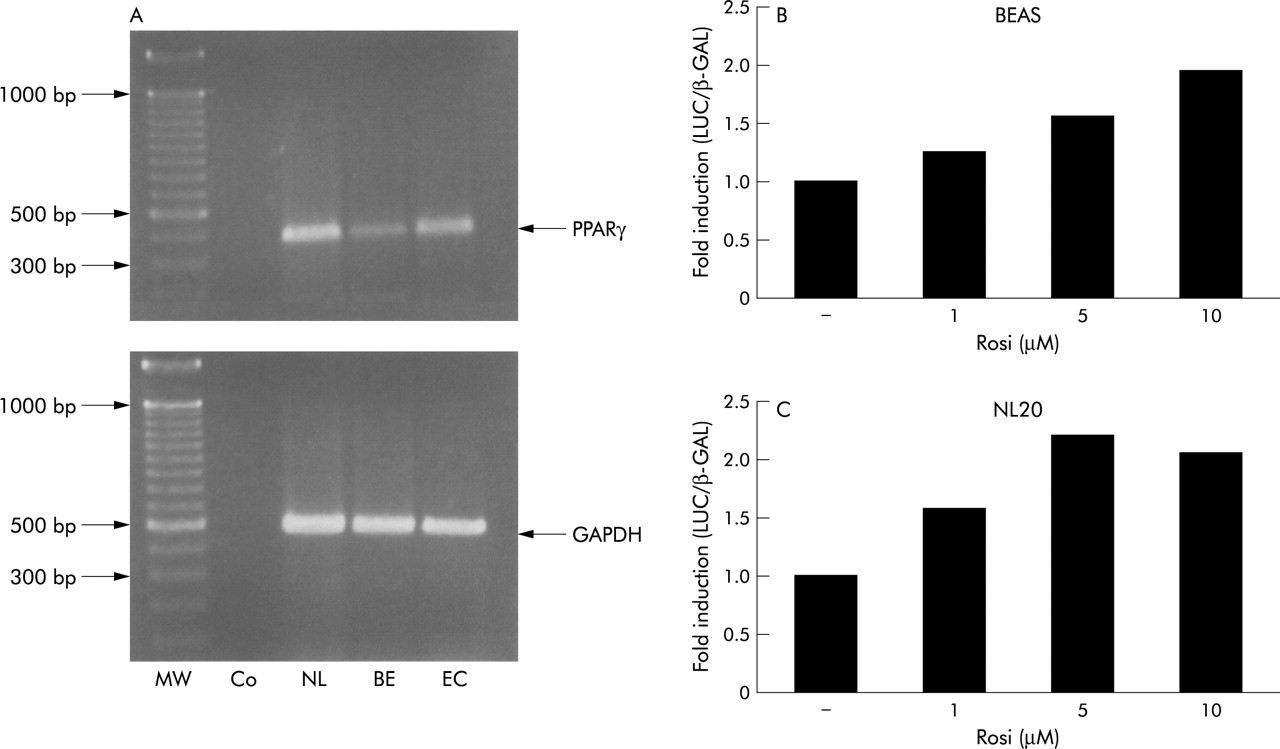
Expression of functionally active PPARγ by human bronchial epithelial cells. (A) RT-PCR of PPARγ RNA in cells of the NL20 (NL) and the BEAS cell line (BE) showing a cDNA fragment of the expected size. Also shown are a DNA ladder (MW), RT-PCR product from endothelial cells as a positive control (EC), and a negative control consisting of RT-PCR reactions lacking reverse transcriptase (Co). (B) and (C) Representative transient transfection assays in (A) BEAS or (B) NL20 cells using a promoter-reporter construct containing three copies of a PPAR response element (PPRE). Transfected cells were stimulated with different concentrations of rosiglitazone for 24 hours before luciferase activity was measured in cell lysates. Data are normalised to the activity of a cotransfected β-galactosidase reporter construct. Three independent experiments yielded similar results.
Activity of PPARγ in human bronchial epithelial cells
To assess the presence of functional endogenous PPARγ we transiently transfected NL20 and BEAS cells with a PPRE-luciferase construct and stimulated cells with increasing amounts of the PPARγ activator rosiglitazone. Luciferase activity was assayed and normalised to β-galactosidase activity of a cotransfected pCMV-β GAL construct. Stimulation with rosiglitazone increased normalised luciferase activity in a concentration dependent manner in both cell lines (fig 1B and C). These results suggest the presence of inducible PPARγ activity in these cell types.
Inhibition of TNF-α or PMA induced MMP-9 gelatinolytic activity by PPARγ activators in human bronchial epithelial cells
Given the presence of functionally active PPARγ in bronchial epithelial cells, we examined the effect of PPARγ activators on MMP activity in these cells. Conditioned media harvested from unstimulated NL20 or BEAS cells exhibited little gelatinolytic activity as determined by substrate zymography. As expected, stimulation with TNF-α significantly induced MMP-9 activity in the supernatant but had no effect on MMP-2 (fig 2A). Incubation of cells with rosiglitazone or pioglitazone significantly reduced this increase in a concentration dependent manner with a maximal reduction to 60 (7)% with 10 μM pioglitazone or to 49 (8)% with 10 μM rosiglitazone (p<0.05 compared with TNF-α -stimulated cells, n=5) in NL20 cells, and to 60 (8)% with 10 μM pioglitazone or to 45 (9)% with 10 μM rosiglitazone (p<0.05 compared with TNF-α stimulated cells, n=3) in BEAS cells. None of the PPARγ activators significantly altered MMP-2 activity (fig 2A and B).

Inhibition of TNF-α induced MMP-9 gelatinolytic activity by PPARγ activators in human bronchial epithelial cells. (A) NL20 (left panel) and BEAS cells (right panel) were stimulated with TNF-α (10 ng/ml) in the absence or presence of the PPARγ activators pioglitazone and rosiglitazone. After 12 hours, cell-free supernatants were subjected to gelatin zymography. (B) Densitometric analysis of MMP-9 gelatinolytic activity in conditioned media from NL20 (left panel; n=5) or BEAS cells (right panel; n=3). The results are expressed as the percentage of TNF-α activated cells (% control). Bars represent mean (SE), *p<0.05.
To examine whether the effects observed depended on the stimulus employed, we performed similar experiments using PMA as an inducer of MMP-9 activity. PMA treatment significantly induced MMP-9 activity, while concomitant treatment with rosiglitazone or pioglitazone diminished this increase significantly in a concentration dependent manner but had no effect on MMP-2 activity (fig 3A and B). Similar results were obtained with primary human bronchial epithelial cells; PMA significantly induced MMP-9 activity and both pioglitazone and rosiglitazone reduced this increase to 47 (3)% and 43 (1)%, respectively (fig 3C).

Inhibition of PMA-induced MMP-9 gelatinolytic activity by PPARγ activators in human bronchial epithelial cells. (A) NL20 (left panel) or BEAS cells (right panel) were stimulated with PMA (10 ng/ml) in the absence or presence of the PPARγ activators pioglitazone and rosiglitazone. After 12 hours, cell-free supernatants were subjected to gelatin zymography. (B) Densitometric analysis of MMP-9 gelatinolytic activity in conditioned media from NL20 (left panel; n=5) or BEAS cells (right panel; n=3). The results are expressed as a percentage of PMA activated cells (% control). Bars represent mean (SE), *p<0.05. (C) Primary human bronchial epithelial cells were stimulated with PMA (5 ng/ml) in the absence or presence of pioglitazone and rosiglitazone (both at 10 μM). After 12 hours, cell-free supernatants were subjected to gelatin zymography (upper panel). Densitometric analysis of MMP-9 gelatinolytic activity of three independent experiments (lower panel). The results are expressed as a percentage of PMA activated cells (% control). Bars represent mean (SE). (D) BEAS cells were treated with TNF-α and PPARγ activators for 12 hours, then again cultured in regular media for 12 hours before additional stimulation with TNF-α in the absence or presence of rosiglitazone (upper panel) or pioglitazone (lower panel) for 12 hours. Gelatin zymography of supernatants taken after first and second stimulation (pretreated with TNF + rosiglitazone or pretreated with TNF + pioglitazone) are shown. Three independent experiments yielded similar results.
Sequential stimulation of cells with PPARγ activators revealed that cells do not become refractory to PPARγ stimulation in inhibiting TNF-α induced MMP-9 activity (fig 3D).
Effect of PPARγ activators on TIMP-1 expression in human bronchial epithelial cells
Since the effects of PPARγ activation on MMP expression did not differ in the two cell lines, further experiments were performed only in BEAS cells. To examine whether PPARγ activators shift the balance of matrix degrading enzymes and their inhibitors towards matrix stabilisation, we measured the release of TIMP-1 (the local inhibitor of MMP-9) after stimulation with PPARγ activators. As shown in fig 4, neither rosiglitazone nor pioglitazone had a significant effect on TIMP-1 protein content in the supernatants from BEAS cells.
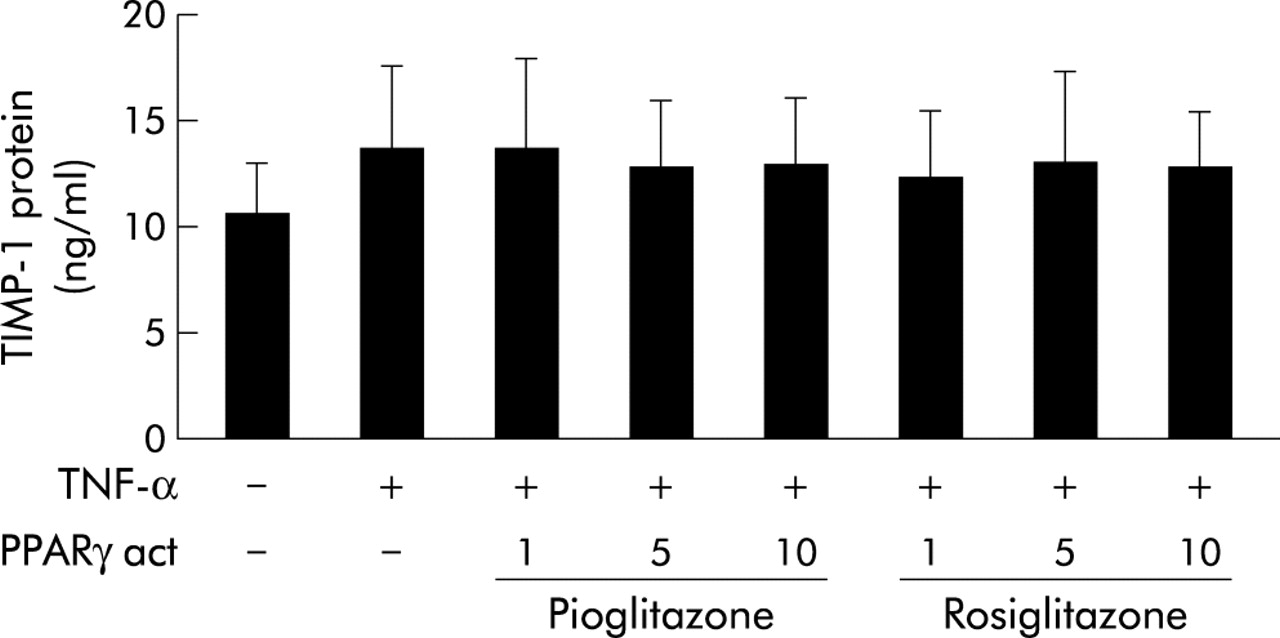
Effect of PPARγ activators on TIMP-1 protein release from human bronchial epithelial cells. BEAS cells were stimulated with TNF-α (10 ng/m) in the absence or presence of PPARγ activating pioglitazone and rosiglitazone. After 12 hours, TIMP-1 protein content in cell-free supernatants was measured by ELISA. The results are expressed as a percentage of TNF-α activated cells (% control). Bars represent mean (SE), n=3.
Effect of PPARγ activators on MMP-9 mRNA expression in human bronchial epithelial cells
To investigate whether the decrease in MMP-9 activity by PPARγ activators resulted from reduced mRNA expression, northern blot analysis was performed after 6 hour stimulation. Both rosiglitazone and pioglitazone markedly reduced the TNF-α induced MMP-9 mRNA content in a concentration dependent manner, but did not affect mRNA levels of the constitutively expressed gene GAPDH (fig 5).
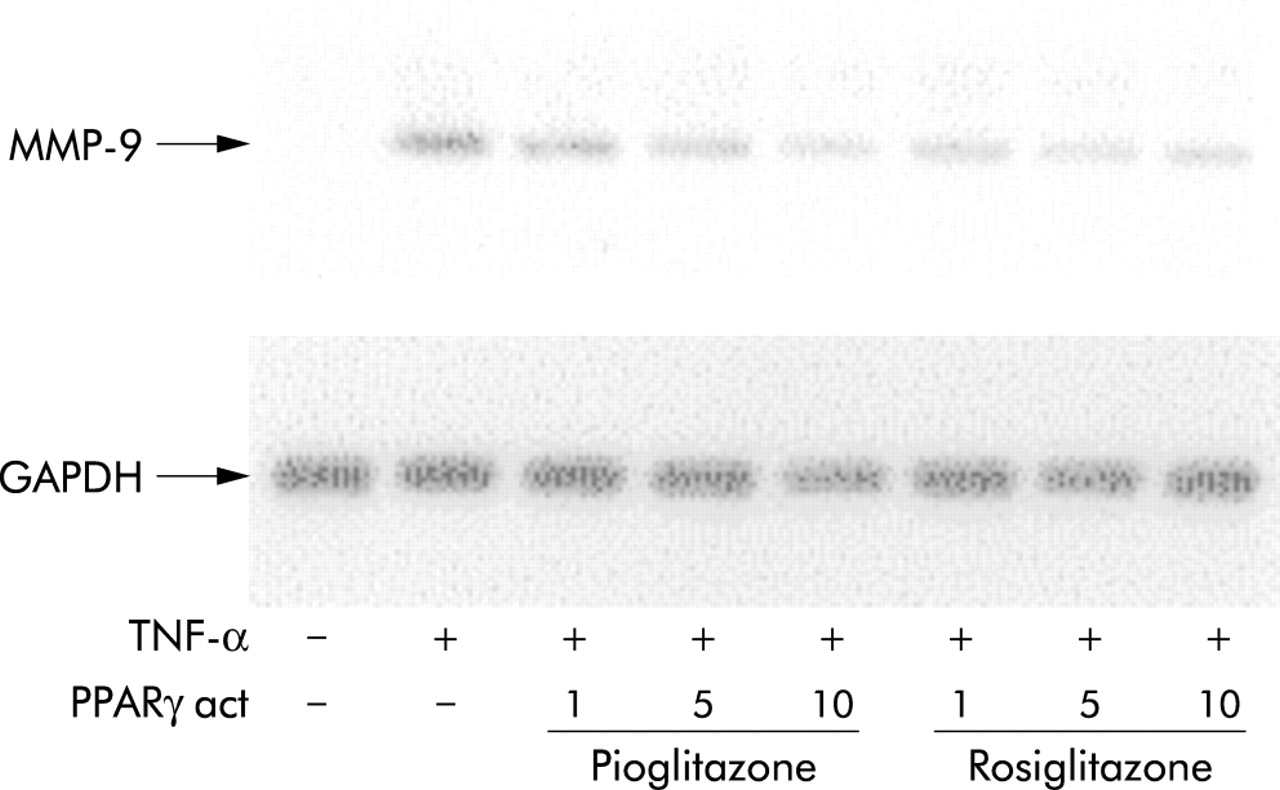
Inhibition of MMP-9 mRNA expression by PPARγ activators in human bronchial epithelial cells. Representative northern blot analysis for MMP-9 expression of BEAS cells treated for 6 hours with TNF-α (10 ng/ml) in the absence or presence of various concentrations of pioglitazone or rosiglitazone. Equal loading of intact RNA is shown by expression of the house keeping gene GAPDH. Three independent experiments yielded similar results.
Effect of PPARγ activators on NF-κB activation in human bronchial epithelial cells
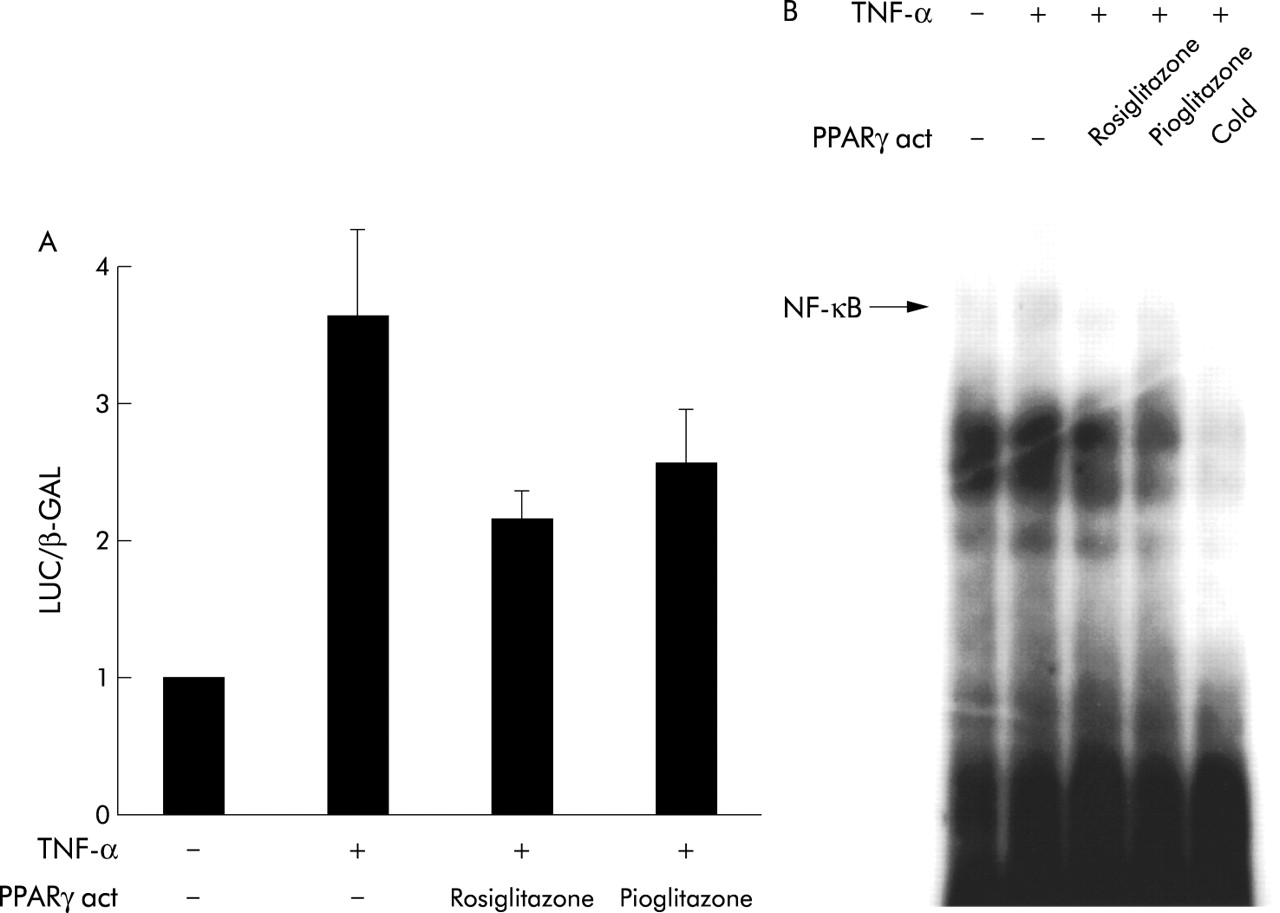
Previous work has shown that TNF-α induced MMP-9 expression in human bronchial epithelial cells is regulated by activation of NF-κB. To assess the effect of PPARγ activation on NF-κB, transient transfection experiments of a reporter construct containing three copies of the MHC class I NF-κB site were used. Stimulation of transfected cells with TNF-α (10 ng/ml) induced a 3.6 (0.6) fold increase in normalised luciferase activity which was reduced to 2.1 (0.2) fold by rosiglitazone and to 2.5 (0.4) fold by pioglitazone (fig 6A). To further investigate whether PPARγ activators inhibit binding of NF-κB transcription factors to the respective NF-κB DNA binding site, we performed electromobility shift assays using oligonucleotides corresponding to the MHCI NF-κB site. As shown in fig 6B, both rosiglitazone and pioglitazone inhibited TNF-α induced binding of the NF-κB proteins to DNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reduction of NF-κB activation by PPARγ activators in human bronchial epithelial cells. (A) BEAS cells were transiently transfected with a promoter-reporter construct containing three copies of a prototypic NF-κB site as well as with a pCMV-β-GAL. Transfected cells were stimulated for 6 hours with TNF-α (10 ng/ml) in the presence or absence of rosiglitazone or pioglitazone (both at 10 μM) before luciferase and β-galactosidase activity was measured in cell lysates. The results are expressed as fold induction of unstimulated cells. Bars represent mean (SE), n=2. (B) Representative EMSA of NF-κB activation in BEAS cells. Cells were stimulated for 2 hours with TNF-α (10 ng/ml) in the presence or absence of rosiglitazone or pioglitazone (both at 10 μM) before nucleic extracts were subjected to EMSA using a prototypic NF-κB site from the MHCI promoter. Specificity of the detected band was determined by competition with addition of excess unlabelled (cold) oligonucleotide. Three independent experiments revealed similar results.
DISCUSSION
This study demonstrates the expression and functional activity of PPARγ in two different human bronchial epithelial cell lines and shows that PPARγ activators inhibit the expression of MMP-9 in these cells through an interaction of PPARγ with the MMP-9 inducing transcription factor NF-κB.
PPARγ expression was originally described in adipocytes and hepatocytes and recent work has demonstrated its presence in inflammatory cells, endothelial cells, and various cells in the intestine. Immunohistochemical staining of normal bronchial tissue revealed PPARγ immunoreactivity in epithelial cells,19 and in vitro experiments suggested the expression of this receptor in lung cancer cells21 and NIH A549 cells. Our study extends the knowledge about PPARγ in the respiratory system by showing PPARγ expression in two different cell lines of human normal bronchial epithelial cells. In addition, the study shows that PPARγ activators are capable of activating endogenous PPARγ in these cells by increasing the activity of a transfected PPRE-luciferase construct.
Stimulation of bronchial epithelial cells with two different PPARγ activators significantly attenuated cytokine induced gelatinolytic activity of MMP-9 but not of MMP-2, another matrix degrading enzyme which is also thought to be implicated in inflammatory airway processes. The ligands employed—rosiglitazone and pioglitazone—are most probably exhibiting their effects through the activation of PPARγ, given that these agents have high binding activities for PPARγ.22,23 However, recent work has suggested that thiazolidinediones might exhibit PPARγ independent effects such as regulation of cell cycle processes in cells of the monocytic lineage.24 We cannot therefore exclude the possibility that similar mechanisms might be involved in the effects investigated in this study.
The action of PPARγ activators on MMP-9 expression is independent of the inducing stimulus since these agents inhibited both TNF-α and PMA induced MMP-9 release, suggesting a direct effect on the expression of this gene. Transient transfection assays and gel shift analyses suggest that PPARγ exhibits its inhibitory effects on MMP-9 expression by limiting TNF-α induced NF-κB activation. In addition, PPARγ seems to limit binding of the NF-κB proteins to the respective DNA binding site. Previous work has shown inhibition of NF-κB activation by PPARγ activators in various cell types such as endothelial cells and monocyte/macrophages.15,25 However, the underlying molecular mechanism of this interaction still needs to be elucidated.
Taken together, these data suggest a novel mechanism to counterbalance the release of matrix degrading MMP-9 from bronchial epithelial cells. PPARγ activation might therefore be useful for preserving the histological airway structure in chronic inflammatory processes, but in vivo experiments and clinical data are needed to determine the biological relevance of MMP-9 inhibition mediated by PPARγ activators.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (MA 2047/2-2, SFB 451) and the Landesschwerpunkt Baden-Württemberg “Die NF-κB -Signalkaskade als Ziel selektiver Interventionsmaβnahmen bei entzündlichen und malignen Krankheitsgeschehen” to Dr Nikolaus Marx.
REFERENCES
Linked Articles
- Airwaves