Article Text

Abstract
Neural lesions occur uncommonly in the gastroenteropancreaticobiliary tract. However, due to the growing number of screening colonoscopy procedures, polypoid neural lesions of the colon are being recognised increasingly and range from benign tumours to high-grade malignant neoplasms. Morphological variability of neural tumours can be wide, although some entities share pathological features, and, as such, these lesions can be diagnostically challenging. We review the spectrum of pathology of neural tumours in the gastroenteropancreaticobiliary tract, with the goal of providing a practical approach for practising surgical pathologists.
- Gastrointestinal tract
- nerve sheath tumours
- neural lesions
- spindle cell neoplasms
Statistics from Altmetric.com
Introduction
Tumours with neural differentiation in the gastrointestinal tract (GIT) represent a conglomerate of relatively common as well as some uncommon neoplasms that are encountered by both general and subspecialist pathologists in their routine practice. Since the GIT is a hollow visceral organ, with a lumen, several of the entities that are discussed here, present within the wall and oftentimes project into the lumen as polypoid lesions, being detected endoscopically as polyps. These tumours can present significant diagnostic challenges both for the pathologist and the clinician, in part because of non-specific and sometimes, overlapping clinical presentations, as well as similarity of histopathological features. Awareness of the wide spectrum of nerve sheath/neural lesions that can manifest in the GIT and pancreaticobiliary tract is crucial for accurate diagnosis and optimal patient management.
This comprehensive review describes the clinical presentation, histological and immunohistochemical features, prognosis and differential diagnosis of common, as well as relatively uncommon, tumours with neural differentiation in the GIT and pancreas.
Table 1 highlights some of the more common neural tumours and the sites of predilection within the GIT.
Most common sites in the gastrointestinal tract (GIT) of neural and neurogenic tumours
Neurofibroma
Clinical features
Neurofibromas are benign nerve sheath tumours that are rare in the GIT and are usually associated with neurofibromatosis type 1 (NF1).1 It has been estimated that up to 25% of patients with NF1 have GI manifestations of the syndrome, with neurofibroma being the most frequent lesion affecting mainly the small bowel, retroperitoneum and less frequently, the colon.2 Visceral neurofibromas may show any of the subtypes commonly observed at peripheral cutaneous and soft tissue sites, including localised, diffuse and plexiform variants.3–7
Gross findings
Localised intraneural neurofibromas are well-defined fusiform or diffuse lesions that appear to be confined to the affected nerve at gross inspection and have a grey to tan cut surface. Plexiform neurofibromas are classically more complex presenting as tortuous or worm-like masses. They involve a nerve plexus or multiple fascicles in a medium-sized to large-sized nerve, with the involved nerves retaining their original configuration and anatomy. Diffuse neurofibromas are typically plaque-like masses that contain diffuse microscopic fat frequently.8
Microscopic findings and immunohistochemistry
Neurofibromas typically contain a mixture of cellular peripheral nerve elements, including Schwann cells, perineurial cells, fibroblasts and variable inflammatory cells, such as mast cells and lymphocytes. Schwann cells represent the primary neoplastic cell component, and they are characterised by wavy nuclear contours and S100 protein expression.9 10 A myxoid background and wavy collagen fibres are characteristic stromal features. A population of CD34-positive cells of unclear histogenesis is also present.11 Other less common morphological findings in neurofibromas include the presence of melanin pigment, metaplastic bone and glandular differentiation.3
Cellular neurofibromas may show moderate cellularity and a more pronounced fascicular growth pattern, but lack the monotonous cytological atypia, coarse chromatin clumping and mitotic activity seen in malignant peripheral nerve sheath tumours (MPNST).11 Neurofibromas with ancient/degenerative change demonstrate degenerative nuclear atypia, characterised by scattered cells with markedly enlarged, hyperchromatic nuclei, often with smudgy chromatin; however, they lack increased cellularity, fascicular growth or mitotic activity.11
Differential diagnosis
The presence of entrapped native axons and relatively small tumour nuclei are helpful features in distinguishing neurofibroma from schwannoma. Fine et al described calretinin staining as a useful immunohistochemical marker to differentiate neurofibromas (only 3 of 42 neurofibromas showed weak, faint staining and the remaining cases did not stain) from conventional schwannomas (24 of 25 schwannomas diffusely expressed calretinin).4
Prognosis
Even though neurofibromas may appear as well-defined masses at gross inspection, they frequently infiltrate into adjacent adipose, muscle and/or viscera. Thus, local recurrences are common after surgical resection.8
Traumatic neuroma
Clinical features
Traumatic neuroma is a non-neoplastic proliferation at a site of nerve transection and it is a well-recognised complication of surgery. It has also been described in bile ducts following biliary surgery, such as after orthotopic liver transplantation5 and cholecystectomy (both laparoscopic and open).6 7 12 Clinically, the leading differential diagnosis typically is cholangiocarcinoma, due to the similarity of presentation, namely jaundice and intermittent symptomatic right upper quadrant pain.7 Rare cases of traumatic neuroma of the colon,13 rectum,14 anus15 and pancreas16 have also been reported. The latency period between surgery and the appearance of the traumatic neuroma may range from a few months to 40 years.5
Microscopic findings
Traumatic neuroma is characterised by a disorganised aggregation of all nerve components, including axons, Schwann cells, perineurial cells and fibroblasts.
Differential diagnosis
It may be difficult to distinguish traumatic neuroma from neurofibroma, but the identification of axon-rich disorganised microfascicles, with a positive neurofilament protein immunohistochemical stain, is a useful distinguishing feature favouring traumatic neuroma.
Schwannoma
Clinical features
Schwannomas are benign neoplasms of Schwann cell origin. Diamaru and colleagues estimated the incidence of myenteric schwannomas as being 2%–6% of all stromal tumours of the GIT.17 They occur most commonly in the stomach,18 19 while schwannomas in the colon and rectum are rare.20–22
Recently, a review of >600 mesenchymal tumours of the colon and rectum showed that the most common location of colonic schwannomas was the caecum, and most of them presented as intraluminal, polypoid masses with mucosal ulceration.19
Microscopic findings
GIT schwannomas are composed almost entirely of well-differentiated Schwann cells, characterised by spindle-shaped cells set in variably myxoid stroma. They are typically unencapsulated lesions, with pushing margins, and are often surrounded by a prominent cuff of lymphoplasmacytic inflammation, including germinal centre formation19 (figure 1). The usual histological features associated with soft tissue schwannomas, namely organisation into Antoni A (Verocay bodies) and B areas, are generally absent.

Oesophageal schwannoma: (A) low-power photomicrograph of an unencapsulated lesion, surrounded by a cuff of lymphoplasmacytic inflammation, with germinal centre formation (A,B). High-power photomicrograph demonstrating spindle-shaped cells set in focally myxoid stroma (C). The lesional cells are strongly and diffusely positive for S100 protein (D).
Rare findings in schwannomas include pseudoglandular structures, epithelioid cells, benign, large cellular palisades resembling neuroblastic rosettes (neuroblastoma-like schwannoma) and lipoblastic differentiation.23–26
Immunohistochemistry and ancillary studies
Schwannomas typically show diffuse, strong S100 protein expression.9 10 Glial fibrillar acid protein (GFAP) is expressed in a subset of schwannomas27 while calretinin4 and SOX1028 are also frequently positive. Rarely, schwannomas may show anomalous expression of cytokeratins.29 Residual epithelial membrane antigen (EMA)-positive and GLUT1-positive perineurial cells can be seen at the periphery of schwannomas and occasionally adjacent to thick-walled blood vessels within the centre of these lesions.30 In contrast to neurofibromas, neurofilament protein staining is usually limited to entrapped axons at the periphery of schwannomas.
Lasota et al showed that GI schwannomas lack alterations in the NF2 gene found in many sporadic, conventional schwannomas from other sites.31
Prognosis
In a study by Miettinen and colleagues, all colorectal schwannomas included behaved in a benign fashion with no evidence of aggressive behaviour.19
Microcystic/reticular schwannoma
Clinical features
Microcystic/reticular schwannoma is a recently described, distinctive morphological variant of schwannoma, with a predilection for viscera, especially the GIT (stomach, small intestine, large intestine), and presentation in an older age group.32–35
Microscopic findings
It is characterised by a striking microcystic and reticular growth pattern, with anastomosing and intersecting strands of plump to slender spindle cells with ill-defined, eosinophilic cytoplasm, set in abundant myxoid, fibrillar, collagenised and/or hyalinised stroma32 (figure 2).

Microcystic/reticular schwannoma: (A) low-power photomicrograph of a lesion characterised by a striking microcystic and reticular growth pattern (A), composed of plump to slender spindle cells with ill-defined, eosinophilic cytoplasm (B), set in hyalinised stroma (C). Immunohistochemistry shows strong and diffuse S100 immunopositivity (D).
Immunohistochemistry
Typically, cases show strong and diffuse S100 immunopositivity and at least focal GFAP immunoreactivity.
Prognosis
All tumours reported behaved in an indolent fashion. Despite the lack of encapsulation and focal permeation into normal tissue at the tumour–normal tissue interface, recurrences have not been described.
Psammomatous melanotic schwannoma
Psammomatous melanotic schwannoma is a rare, distinctive, potentially malignant neoplasm that has been encountered as a primary GIT lesion.
Microscopic findings and immunohistochemistry
This tumour is characterised by mainly epithelioid cells with focal spindle cells and areas of palisading and pseudorosette formation around vessels. The nuclei of tumour cells are uniform with minimal pleomorphism, and some contain small nucleoli and several contain nuclear pseudo-inclusions. The epithelioid cells have abundant eosinophilic cytoplasm and several contain melanin pigment, with variably sized nuclei and a striking accumulation of melanin pigment in neoplastic cells, as well as in associated melanophages.36 Several multinucleated giant cells with a ‘mulberry’ appearance are also present (figure 3). The presence of prominent psammoma bodies is seen frequently (in about 50% of cases) in those tumours associated with Carney complex (characterised also by skin pigmentary abnormalities, myxomas and endocrine tumours or overactivity; pathogenic variants in PRKAR1A are causative).37 38 However, sporadic forms also occur and contain psammoma bodies as well. Occasional lipid-laden macrophages are interspersed between tumour cells and sometimes fat can be encountered. The mitotic count is extremely low and found after exhaustive searching. The stroma varies from fibrillar to oedematous to microcystic. There is no evidence of cytological atypia or necrosis. The tumour is diffusely positive for S100, and focally for HMB45 and Melan A.

Low-power photomicrograph depicting a psammomatous melanotic schwannoma (A), characterised by uniform epithelioid cells with minimal pleomorphism. Several multinucleated giant cells with a ‘mulberry’ appearance are present (B). Many lesional cells contain melanin pigment (C) and prominent psammoma bodies are seen (D).
Differential diagnosis
The differential diagnosis includes melanoma, gastrointestinal stromal tumour (GIST), pigmented neuroendocrine tumour and epithelioid leiomyoma. The absence of malignant features separates this lesion from melanoma while the immunophenotype of the other lesions is characteristic.
Perineurioma
Clinical features
Perineurioma is an uncommon and under-recognised benign neoplasm, defined as a peripheral nerve sheath tumour composed exclusively of neoplastic perineurial cells that demonstrate ultrastructural and immunohistochemical features similar to those of their normal cell counterpart.39 In contrast to other peripheral nerve sheath tumours, perineuriomas have not been found to be associated with neurofibromatosis. They mainly affect adults. Perineuriomas include soft tissue, intraneural and sclerosing variants.40–43
Perineuriomas of the GIT occur most commonly in the left and distal colon,40 44 45 but may also occur in the small intestine.40 Mucosal perineuriomas are typically asymptomatic, small, sessile, intramucosal lesions (also called benign fibroblastic polyps),46 which may be detected during screening colonoscopy. In contrast, perineuriomas of soft tissue type are rare submucosal or intramural masses.40
Microscopic findings
Mucosal perineuriomas are histologically distinctive lesions that entrap native crypts and are composed of uniform, cytologically bland spindle cells, with ovoid to tapered nuclei and indistinct eosinophilic cytoplasm, lying in a delicate collagenous background40 (figure 4). Many lesions also show a focal pericryptal whorling growth pattern. Hyperplastic/serrated crypts represent a common finding in colonic intramucosal perineuriomas, being reported in 50%–100% of cases.40 46–48 It is unclear whether the perineurial component is reactive or neoplastic in origin, although the presence of 22q deletions in perineuriomas supports a neoplastic origin for this entity.43 The extent of the perineurial proliferation among colonic crypts is variable. Indeed, some cases may even be missed as they contain only subtle and limited amounts of perineurial cells.48 49 Hornick and Fletcher described two submucosal perineuriomas of soft tissue type, morphologically similar to their soft tissue counterparts, being composed of spindle cells with tapered nuclei and elongated bipolar cytoplasmic processes.40

Perineurioma of the sigmoid colon, comprising a spindle cell lesion in the lamina propria, with overlying hyperplastic colonic crypts (A). The lesional cells are uniform, cytologically bland spindle cells, with ovoid to tapered nuclei and indistinct eosinophilic cytoplasm (B). Immunohistochemistry shows that the spindle cells are positive for claudin-1 (C) (courtesy of Professor Christopher Fletcher, Brigham and Women’s Hospital, Boston, USA) and negative for S100 and smooth muscle actin (neither shown). GLUT1 (D) (courtesy of Professor Ivan Chebib, Massachusetts General Hospital, Boston, USA) is also positive in perineuriomas (H&E of same lesion in inset of image D).
Immunohistochemistry and ancillary studies
Ancillary studies are generally required for the diagnosis of perineurioma. EMA stains the majority, typically in a membranous fashion (may be extremely limited or faint, depending on the antibody dilutions and incubation periods used).50 Claudin-1 and GLUT1 are typically positive, both being regarded as highly sensitive and relatively specific for perineurioma.51 52 It has been recommended that at least two immunohistochemical markers of perineurial differentiation should be used to reach an accurate diagnosis.46
Agaimy and colleagues analysed a series of colonic mucosal perineuriomas with serrated crypts for mutations in the serine/threonine kinase, BRAF 48 (hyperplastic polyps and sessile-serrated polyps commonly harbour mutations in BRAF 53 54). In their study, 63% harboured a pV600E BRAF point mutation, indicating that the serrated epithelium is a neoplastic rather than a reactive phenomenon.48 An identical molecular alteration was subsequently found by Pai et al and Pettus et al, with the latter study suggesting that the BRAF mutation is located in the epithelium rather than in the perineurial stromal cells.47 55
Differential diagnosis
The differential diagnosis of perineuriomas of the GIT depends on whether the lesion is intramucosal or submucosal. For colorectal intramucosal perineuriomas, the differential diagnosis includes neurofibroma,1 ganglioneuroma (GN),56 57 schwannoma, neuroma, Schwann cell hamartoma,58 benign epithelioid nerve sheath tumour59 and leiomyoma of the muscularis mucosae.60 For submucosal perineuriomas, the primary differential diagnoses are schwannomas19 and GISTs.61 62
Ganglioneuroma
Clinical features
Ganglioneuromas (GNs) are benign lesions, composed of ganglion cells, nerve fibres and Schwann cells.58 They occur typically in the retroperitoneum and posterior mediastinum, but may also arise in the GIT, where they most commonly involve the colon and rectum.57 Colorectal GNs may present as solitary polypoid lesions, multiple polyps (ganglioneuromatous polyposis) or as diffuse ganglioneuromatosis (DG).57 Gallbladder involvement by ganglioneuromatosis has also been reported.63–65
Solitary polypoid GNs, the most common form of colorectal GNs, are usually sporadic, and their presence does not suggest a genetic disorder.57 In ganglioneuromatous polyposis, there are typically >20 sessile or pedunculated mucosal and/or submucosal lesions.57 Associated peripheral and GI lipomas are frequently reported. Ganglioneuromatous polyposis may be associated with PTEN hamartomatous tumour syndrome (unified syndrome including Cowden disease and Bannayan–Ruvalcaba–Riley syndrome).66 The polyps may be microscopically indistinguishable from polypoid GNs. DG is a disseminated, nodular, intramural or transmural proliferation of neural elements that involves the enteric plexuses.57 It may exist as an isolated finding, but is often associated with NF1 or multiple endocrine neoplasia syndrome type IIb (MEN-IIb).
Microscopic findings and immunohistochemistry
Histologically, GNs have ill-defined margins and generally show entrapment of native colonic crypts. They are composed of an admixture of S100 protein-positive spindled Schwann cells, neurofilament protein-positive ganglion cells and axons, set in a fibrillar matrix (figure 5). Eosinophils may be frequent.

Appendiceal ganglioneuroma comprising an ill-defined mucosal lesion with entrapment of native colonic crypts (A). It is composed of an admixture of spindled Schwann cells (B) and ganglion cells (C,D).
Significance
It is possible that the development of ganglioneuromatosis may be due to defective tumour suppressor gene signalling. Cowden syndrome, NF1 and MEN-IIb all have tumour suppressor gene abnormalities (PTEN, NF1 and RET, respectively).56 Additionally, these tumour suppressor genes all have overlapping signalling via the Akt and mTOR signal transduction pathways. Thus, GN formation in Cowden syndrome, NF1 and MEN-IIb may result from inappropriate signalling along the Akt–mTOR pathway.56
It is important for pathologists to recognise these lesions so that opportunities to identify patients and families with tumour syndromes are not missed.
Gangliocytic paraganglioma
Gangliocytic paraganglioma (GP) is a rare, typically benign tumour of the GIT. The name is derived from its unique histological features showing neuroectodermal (neurosustentacular or Schwannian and neuronal) and neuroendocrine differentiation. The lesion is composed of three types of cells: spindle cells, ganglion-like cells and epithelioid cells. Many theories have been offered to explain the histogenesis of GP, but so far no clear explanation has been formed.
Clinical features
GPs occur nearly exclusively in the second portion of the duodenum, particularly at the ampulla of Vater or in the periampullary region. They have also been reported in the third and fourth parts of the duodenum, at the minor papilla, with rare cases reported in the jejunum, pylorus, oesophagus, pancreas and appendix.67–69
Gross findings
GPs are characteristically detected incidentally at endoscopy as submucosal tumours and assume the form of circumscribed, pedunculated or sessile lesions.
Microscopic findings
Histopathologically, GPs are unencapsulated and infiltrative tumours, consisting of three cell components with different types of differentiation: columnar or polymorphic epithelioid cells with a nested or ribbon-like arrangement, spindle cells with a cord-like arrangement and scattered ganglion-like cells (figure 6). The ratio of these three cell components differs among patients, although the ganglion cells are typically less numerous and dispersed singly or in small groups (two to three cells), surrounded by spindle cells.

Gangliocytic paraganglioma: low-power photomicrograph of an unencapsulated tumour (A), consisting of columnar epithelioid cells with a nested arrangement (B), spindle cells (C) and scattered ganglion-like cells (D). Immunohistochemically, the epithelioid cells stain positively with a wide variety of antigens, including chromogranin (E), synaptophysin (F) and somatostatin (G). The spindle cells stain strongly with S100 protein (H).
Immunohistochemistry
Immunohistochemically, the epithelioid cells stain positively with a wide variety of antigens, including cytokeratin, synaptophysin, chromogranin, neuron-specific enolase (NSE), pancreatic polypeptide and somatostatin. The finding of keratin immunoexpression in epithelioid cells of GPs supports neuroendocrine differentiation. The spindle cells almost always stain strongly with S100 protein, but can also stain with NSE, neurofilament protein(NFP) and vimentin, suggesting neural, particularly Schwannian, differentiation. In addition, the cytological and immunohistochemical features of spindle cells are very similar to those of sustentacular cells observed in the adrenal medulla as well as in paraganglioma and phaeochromocytoma. The ganglion cells are frequently positive with NSE, synaptophysin and NFP.70 71
Prognosis
GPs generally behave in a benign fashion, although instances of recurrence and/or lymph node metastasis have been described.67 72 73 To date, histological features predicting malignant potential have not been defined, although the presence of nuclear pleomorphism, mitotic activity and infiltrative margins raises concern for aggressive behaviour.74
Mucosal Schwann cell hamartoma
Mucosal Schwann cell hamartoma (MSCH) was first described by Gibson and Hornick in 2009 as a sporadic colonic polypoid lesion of neural origin in an attempt to distinguish these lesions from true ‘neurofibromas’ and ‘neuromas’.58
Clinical features
MSCH are usually asymptomatic and identified incidentally on screening colonoscopy, predominantly in the distal colon.58 75 76 Recently, there has been a case report describing MSCH outside the colon, in the gastric antrum.77 These lesions are most commonly detected in middle-aged to elderly patients with a slight female predominance.58 76 78 79
Gross and microscopic findings
MSCHs are typically small, sessile polyps a few millimetres in diameter, with a reported mean size of 2.5 mm.58 76
Histologically, these polyps are composed of a poorly circumscribed, ill-defined mucosal proliferation of uniform, bland-appearing spindle cells (figure 7). The spindle cells exhibit wavy, elongated or tapering nuclei with inconspicuous nucleoli, abundant eosinophilic cytoplasm and indistinct cell borders. The lesional cells entrap colonic crypts and have an irregular border with the adjacent uninvolved lamina propria. Cytological atypia, pleomorphism and mitotic activity are not seen.58 75 76
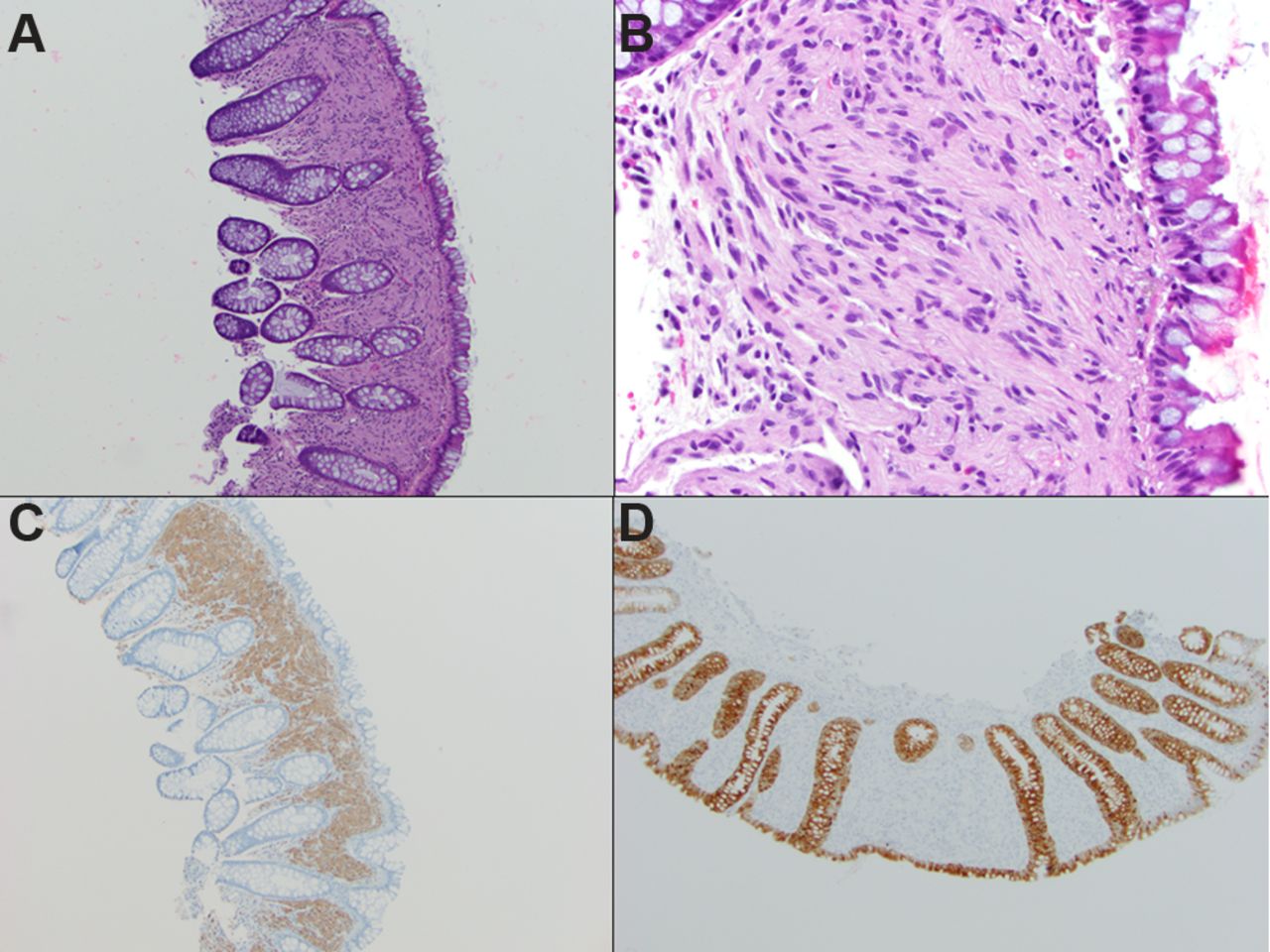
Schwann cell hamartoma: (A) low-power photomicrograph depicting an ill-defined mucosal proliferation of uniform, bland-appearing spindle cells entrapping the colonic crypts (H&E, ×100). (B) High-power photomicrograph highlights wavy, elongated nuclei with inconspicuous nucleoli, abundant eosinophilic cytoplasm and indistinct cell borders. The lesional cells are diffusely positive for S100 protein (C, ×100) and negative for epithelial membrane antigen (D, ×100).
Immunohistochemistry
The lesional cells show strong and diffuse nuclear as well as cytoplasmic staining for S100 protein. Immunohistochemical stains for EMA, GFAP, keratins, CD34, smooth muscle actin (SMA) and c-kit are typically negative.58 75 76 78 79
Treatment and prognosis
MSCH appears to be benign, and recurrences and malignant behaviour have not been documented in the limited studies with clinical outcome available in the literature. To date, they have shown no association with specific inherited syndromes such as NF1, MEN-IIb or PTEN hamartoma tumour syndrome.58 75 76 78 79
Differential diagnosis
The histological differential diagnosis includes other neural lesions such as benign epithelioid nerve sheath tumour, perineuriomas, mucosal neuromas, neurofibromas, intramucosal schwannomas and polypoid GNs, and the features distinguishing these histological mimics from MSCH are described elsewhere in this paper. Lastly, GIST and smooth muscle tumours may be considered in the differential diagnosis, but usually either do not involve, or are not limited to, the mucosa. GISTs show immunoreactivity for DOG1 and CD117, while smooth muscle tumours are diffusely positive for muscle-specific markers such as SMA, caldesmon and desmin, while being negative for S100 protein.
Mucosal benign epithelioid nerve sheath tumour
Another relatively recently characterised colorectal neural polyp, mucosal benign epithelioid nerve sheath tumour (MBENST), was first described by Lewin et al in their series of six small colonic polyps.59 These are rare tumours and their classification as either schwannomas or neurofibromas remains unclear because of differing clinicopathological features between these entities and many authors favour the non-descript term ‘benign epithelioid peripheral nerve sheath tumours’ (BEPNSTs) to describe these lesions.59 80
Clinical features
MBEPNSTs usually present as incidental small colonic polyps, mostly in the left side of the colon. These can occur in a wide age range of patients with a reported mean age of 58.6 years. In the limited literature about these lesions, none of the patients had a known history of neurofibromatosis.59
Gross and microscopic findings
Grossly, they present as small polyps, usually varying from 0.2 to 1 cm in greatest dimension.
On routine H&E staining, MBENST mostly display pushing or infiltrative borders, while on rare occasions they can be encapsulated. Although the epicentres of these lesions are mostly in the lamina propria, they can extend into the superficial submucosa and can sometimes have a substantial submucosal component. In fact, in the series by Lewin and colleagues, one tumour was confined entirely to the submucosa.58 59 75
MBENSTs exhibit collections of bland, spindled to mostly epithelioid cells with abundant eosinophilic to clear cytoplasm arranged in nests and whorls (figure 8). The nuclei are uniform, bland and round to oval with pale nuclear pseudoinclusions. These lesions do not exhibit Verocay bodies. No mitoses or nuclear pleomorphism are seen. The overlying crypt epithelium may show reactive epithelial changes along with mild crypt distortion.59

Mucosal benign epithelioid nerve sheath tumour: (A) low-power photomicrograph highlighting mucosal collections of bland, spindled to mostly epithelioid cells arranged in whorls (H&E, ×100). (B) High-power photomicrograph exhibits bland, uniform, oval nuclei with pale nuclear pseudoinclusions (marked by arrow) and abundant eosinophilic to clear cytoplasm (H&E, ×400). The lesional cells are diffusely positive for SOX10 (C, ×100) and S100 protein (D, ×100).
Immunohistochemistry
The lesional cells are diffusely positive for S100 protein and SOX10, and are negative for keratin markers, GFAP, HMB45, Melan A, c-kit and calretinin.59 80
Treatment and prognosis
In the limited literature about these lesions, all the encountered lesions were small and treated by polypectomy. All patients have remained asymptomatic with no signs of recurrence of the lesions or any metastatic disease. Hence, the pathologist should recognise these as benign lesions. Whether these lesions are related to Schwann cell hamartomas, represent distinct entities or fall within a spectrum of benign intramucosal Schwann cell proliferations is not entirely clear, but the presence of a submucosal component is certainly different from MSCH.58 59 75
Differential diagnosis
A wide variety of lesions are included in the differential diagnosis of these BEPNSTs in mucosal sites. The histological differential diagnosis includes other neural lesions such as Schwann cell hamartoma, mucosal neuromas, neurofibromas, intramucosal schwannomas and polypoid GNs, and the distinguishing features of these lesions have been described elsewhere in this paper. BEPNSTs typically have a bland cytomorphology, and this, coupled with the lack of expression of epithelial markers and other specific melanoma markers such as HMB45 and Melan A on immunohistochemistry, can easily help eliminate carcinomas and carcinoid tumours as well as malignant melanomas (MM) from the differential diagnosis. In addition, carcinoma and MM typically show cellular and nuclear pleomorphism, along with mitotic figures. GISTs can have an epithelioid appearance with rounded cells with clear or lightly eosinophilic cytoplasm and may pursue a malignant course and hence needs to be distinguished from BEPNSTs. GISTs typically lack prominent intranuclear inclusions and are positive for DOG1 and CD117 while being negative for S100, unlike BEPNSTs. Leiomyomas may also be encountered arising in association with the muscularis mucosae of the colon. However, they display brightly eosinophilic cytoplasm and blunt-ended nuclei. By immunohistochemistry, leiomyomas express desmin, calponin and caldesmon but lack S100 protein.
Granular cell tumour
Granular cell tumours (GCTs) are predominantly benign tumours that are thought to be derived from Schwann cells. Although most commonly found in the subcutaneous tissue and oral cavity, especially the tongue, they may occur throughout the GIT where they are much less common.75 81 82
Clinical features
GCTs in the GIT are predominantly solitary and are most commonly observed in the oesophagus, followed by colorectum and then stomach.81 The majority of colorectal GCTs involve the right colon.82 83 Occasionally, one may encounter multiple lesions arising in different portions of the GIT, either synchronously or metachronously, although multiplicity is very rare. GCTs show a male preponderance with a male-to-female ratio of 2.2:1 and a mean age of 49 years. A recent study showed that gastric and colorectal GCTs (mean size of 0.75 and 0.6 cm, respectively) were significantly larger than oesophageal tumours (mean size of 0.27 cm).75 81
Gross and microscopic findings
GCTs are composed of either plump, polygonal cells or spindled cells, with abundant eosinophilic, granular cytoplasm (figure 9) containing numerous periodic acid–Schiff (PAS)-positive, diastase-resistant granules. The nuclei may be small and uniform or large with open chromatin. Rarely, nuclear pleomorphism is prominent. The cells are typically arranged in nests with intervening fibrous septae or sheets. The edges of the tumour can be either infiltrating or circumscribed, and there is sometimes a peritumoural lymphoid cuff. A recent study reported that colorectal and gastric GCTs showed a more infiltrative growth pattern and peritumoural lymphoid cuffs compared with oesophageal tumours. Hyalinisation or calcification or both occur sometimes.75 81–83

Low-power photomicrograph depicting (A) oesophageal granular cell tumour (GCT) seen mainly in the lamina propria and abutting the overlying squamous epithelium (H&E, ×100); (B) submucosal colonic GCT with overlying reactive epithelial changes (H&E, ×40). (C) Medium-power photomicrograph depicts sheets of plump, polygonal as well as spindled cells, with intervening fibrous septae in GCT. The cells show abundant eosinophilic, granular cytoplasm and small, uniform nuclei (H&E, ×200). The lesional cells are diffusely positive for S100 protein (D, x100), while the overlying squamous epithelium is negative.
In the oesophagus, GCTs are situated mainly in the lamina propria, and abut the overlying squamous epithelium, which often shows pseudoepitheliomatous hyperplasia, that can mimic squamous cell carcinoma in superficial endoscopic mucosal biopsies—a pitfall which pathologists need to be aware of. In the colon, they are mainly found in the submucosa and commonly extend into the base of the mucosa. The overlying mucosa may show regenerative epithelial changes that can mimic an adenoma—another pitfall to keep in mind. Although invasive GCT is rare, it has been observed in the GIT.75 81–83
Immunohistochemistry
GCTs show diffuse strong positivity for S100 protein and SOX10. Other markers which are variably reported positive include SOX10, NSE, panmacrophage marker CD68 and CD56. Inhibin-α positivity is seen in about half of cases and is seen more commonly in colonic GCTs than oesophageal tumours. The cytoplasmic granules are positive for PAS and diastase-resistant.75 81–83
Treatment and prognosis
GCT typically behaves in a clinically benign fashion, and local excision is considered adequate therapy. However, regrowth of the lesion may occur if incompletely excised, and hence follow-up may be warranted.
Fanburg-Smith et al in their series of soft tissue GCTs evaluated six histological features: necrosis, spindling of tumour cells, vesicular nuclei with large nucleoli, increased mitotic rate (>2 mitoses per 10 high-power fields (HPFs) at 200× magnification), high nucleus-to-cytoplasm (N:C) ratio and pleomorphism. They graded the soft tissue GCTs histologically into benign (none of the above criteria or focal pleomorphism), atypical (1–2 criteria) and malignant (3–6 criteria).84 However, it is not entirely clear whether these criteria are useful in GCTs of the GIT, although it may seem logical to infer that these criteria would hold true within other sites as well. Subsequently, Nasser et al proposed another diagnostic criteria based on the presence of necrosis (whether single cell or zonal) and/or mitoses, and designated tumours with none of the above two features as benign, and tumours demonstrating at least one feature as GCTs with uncertain malignant potential. They considered metastases as the only denominator of malignant cases.85 86
Differential diagnosis
The histological differential diagnosis includes some other neural lesions such as BEPNST and SCH. The distinguishing features of these lesions are discussed elsewhere in this paper. Epithelioid GISTs are also in the differential, and the lesional cells have abundant cytoplasm with perinuclear halos, variable mitotic activity and are DOG1 and c-kit positive, while being negative for S100, SOX10 and CD68, unlike GCTs. Xanthomas are aggregates of lipid-laden histiocytes with foamy cytoplasm and central bland nuclei. They can mimic GCT histologically and are CD68 positive like GCT, although they are negative for markers of neural differentiation such as S100 and SOX10. Although rare, signet ring cell carcinoma is an important differential. These tumours show cells with cytoplasmic mucin vacuoles with eccentric, hyperchromatic and pleomorphic nuclei and are keratin positive, while being negative for S100 and SOX10.
Fibrous obliteration of the appendiceal lumen
The obliterated appendix has been described in the literature as a conglomerate of varied terms including neuroma, neuronal hyperplasia, neuromatosis of the appendix, neurogenic appendicitis and fibrous involution of the appendix. A consensus among most authors is that the neurogenic tissue is a vital part of this lesion, and the finding of lesions with a predominantly neural composition led to this alternative designation of ‘appendiceal neuroma’ or ‘neuronal hyperplasia’.87 88
Clinical features
This lesion is seen in over half of autopsy specimens and in approximately one-third to one-fourth of excised appendices. It is usually an incidental finding. Although the occurrence of this lesion varies from young age to senescence, the frequency of occurrence increases with patient age. There is no age difference between subjects with focal obliteration versus complete obliteration.88
Gross and microscopic findings
Grossly, typically no tumour nodule is seen. The appendix may appear white and narrow with luminal obliteration in the lesional area compared with adjacent normal appendix. The distal end of the appendix is usually affected, but the whole appendix may be involved gradually.
Microscopically, different architectural patterns have been described. The most common is the central obliterative lesion which shows an effacement of the mucosa and submucosa with loss of lymphoid follicles (figure 10). It is characterised by a loose proliferation of spindle cells that may expand the lamina propria usually in a fibromyxoid background, frequently with entrapped fat. The lesional cells include Schwann cells, non-myelinated nerve fibres and fibroblasts. Admixed inflammatory cells including mast cells, and eosinophils, as well as scattered neuroendocrine cells may also be present. The mucosa proximal to the obliterated portion features varying degrees of nerve proliferation. The intramucosal neuroma pattern shows expansion of the lamina propria by a loose spindle cell proliferation that widely separates the crypts; however, it does not obliterate the lumen. Lastly, a nodular appendiceal neuroma has been described to be characterised by an unencapsulated but well-circumscribed nodular accentuation of the central obliterative process.87–89
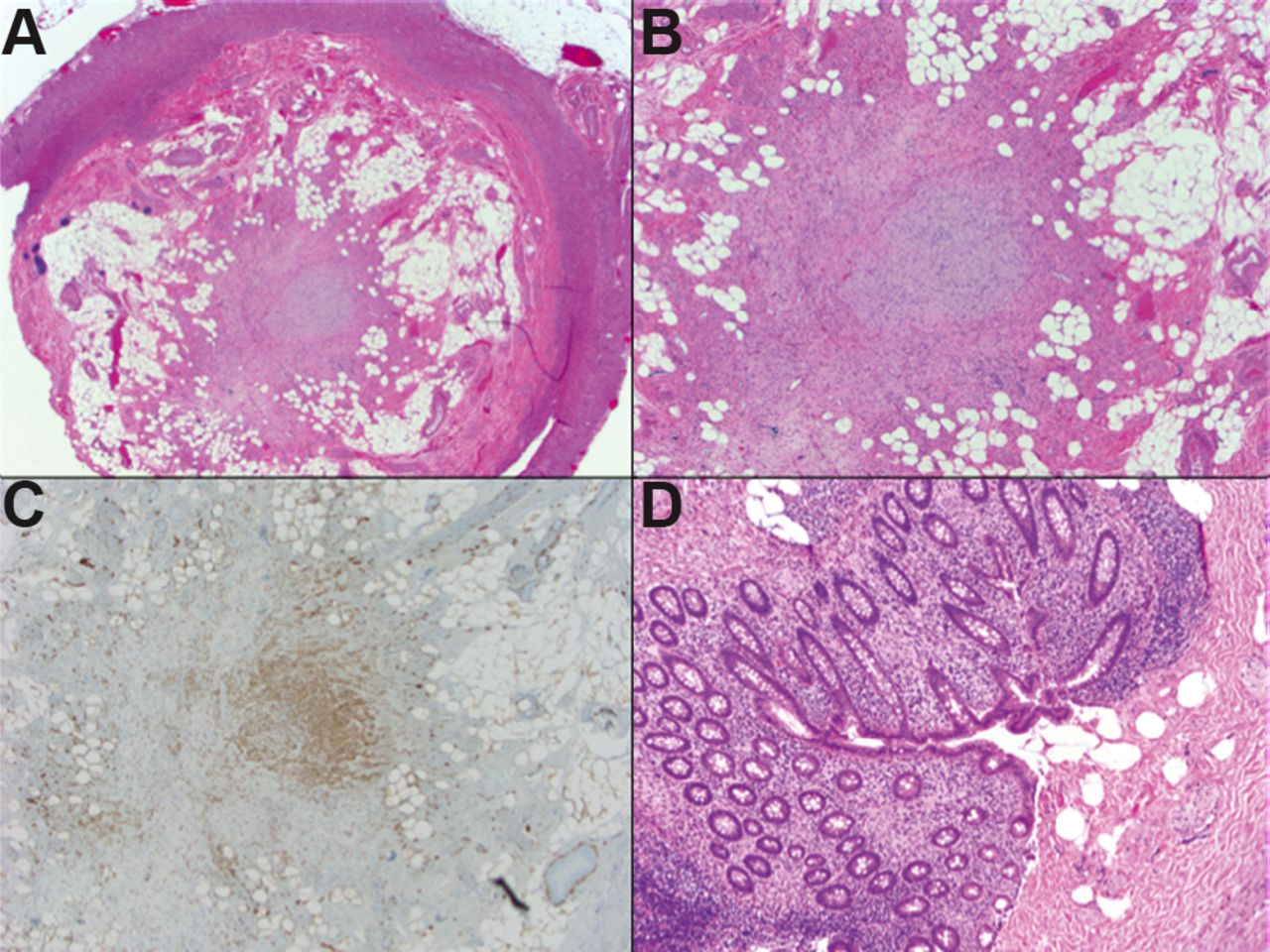
Fibrous obliteration of the appendix: (A) Central obliterative lesion with effacement of the mucosa and submucosa and loss of lymphoid follicles (H&E, ×20). (B) Photomicrograph highlights a loose proliferation of spindle cells in fibromyxoid background, with entrapped fat (H&E, ×40). (C) The lesional cells are positive for S100 protein (C, ×40). (D) The intramucosal neuroma pattern showing a loose spindle cell proliferation in the lamina propria (S100 positive, not depicted in these micrographs) that does not obliterate the lumen (H&E, ×100).
Immunohistochemistry
Immunohistochemical staining shows a mixed population of S100 protein positive Schwann cells, NSE-reactive spindle cells corresponding to axons and CD34-positive admixed fibroblasts.87 88
Treatment and prognosis
This lesion is thought to be a reactive process, either as a normal process of ageing or as a consequence of prior attacks of inflammation such as prior acute appendicitis, with subsequent phases of growth, involution and fibrosis. The common occurrence of these lesions in appendices with ruptured diverticula further supports the dictum that this is a reactive process.87–90 Alternately, some authors have postulated that neural proliferation is common in appendiceal diverticular disease and may serve as a histopathological marker for the presence of coexisting diverticular disease.89
Differential diagnosis
The differential diagnosis may include carcinoid tumour which also occurs in the distal appendix. However, carcinoid tumour is usually associated with a gross nodule or thickening of the appendiceal wall. Histologically, neoplastic cells are seen arranged in nests, sheets or insular pattern and are positive for neuroendocrine markers while being negative for S100.
Gastrointestinal neuroectodermal tumour (clear cell sarcoma-like tumour of the GIT)
Malignant gastrointestinal neuroectodermal tumour (GNET) was first reported by Zambrano et al as ‘osteoclast-rich tumour of the gastrointestinal tract with features resembling clear cell sarcoma (CCS) of soft parts’ in their series of six distinctive tumours of the GIT.91 These tumours shared some features with CCS of soft parts and showed prominent osteoclast-type multinucleated giant cells, but lacked evidence of melanocytic differentiation. Given the absence of immunophenotypic and ultrastructural evidence of melanocytic differentiation in these tumours, they have also been referred to as ‘clear cell sarcoma like tumour of the gastrointestinal tract’.92 93 Because these tumours exhibit a neural line of differentiation, Stockman and colleagues designated these as GNET.91–98
Clinical features
The usual clinical presentation includes abdominal pain, intestinal obstruction, weight loss and an abdominal mass. Some patients have metastatic disease at the time of initial diagnosis. These tumours most commonly are seen in the small intestine, followed by the stomach and the colon. Overall, GNETs can occur over a wide age range from 10 to 81 years; however, they tend to occur mostly in young to middle-aged adults.91–93 97 98
Gross and microscopic findings
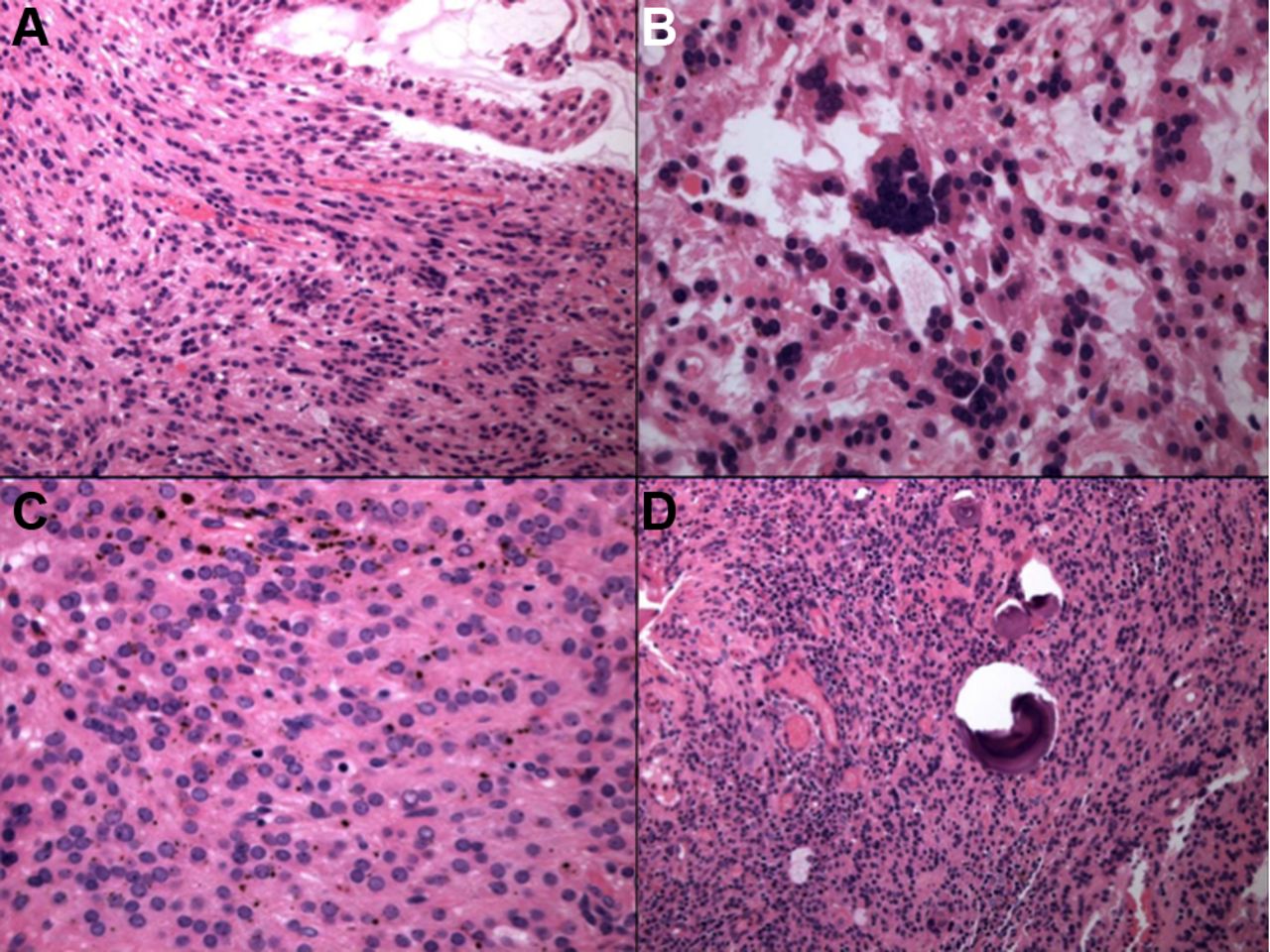
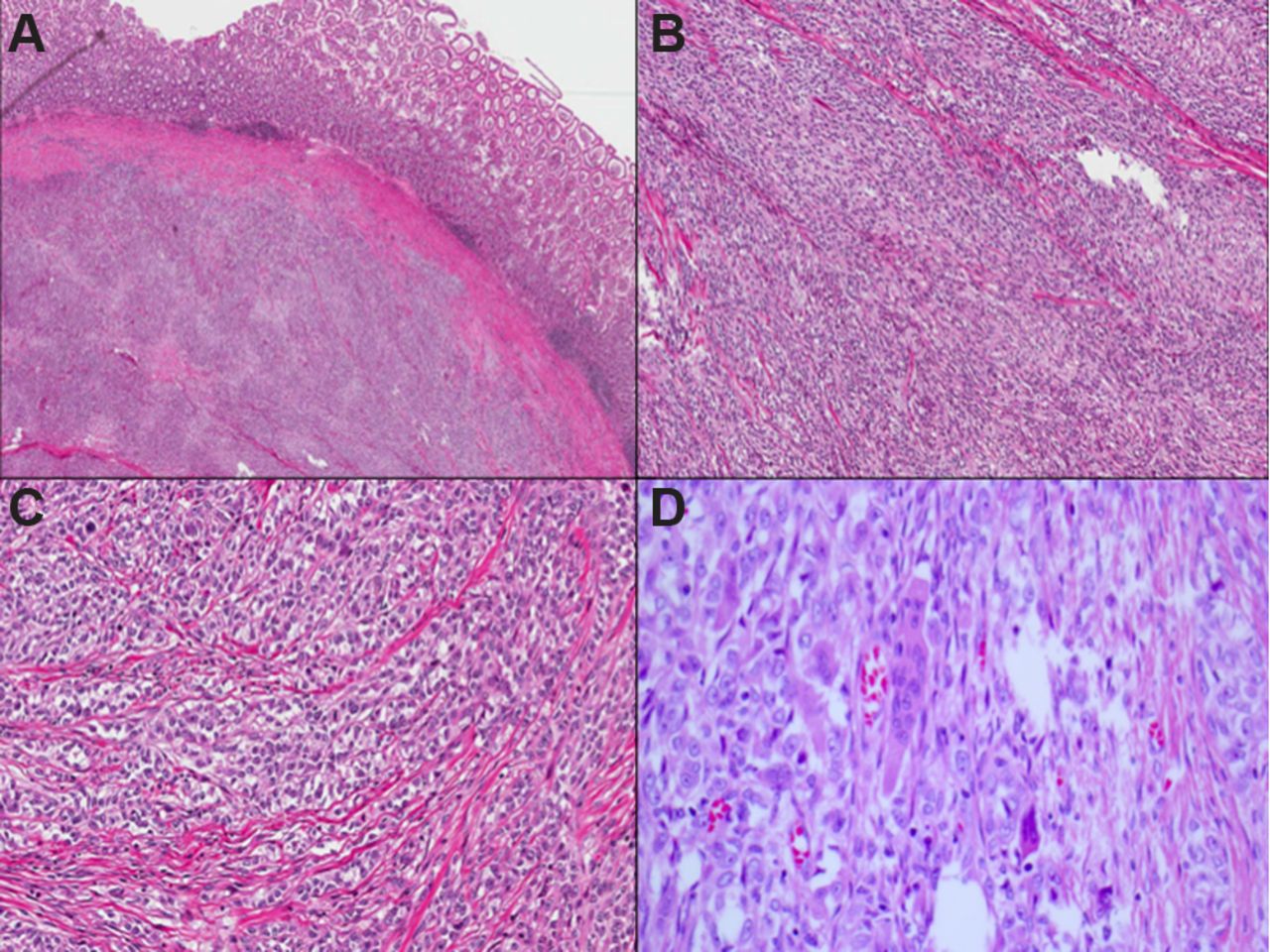
GNET is typically centred within the muscularis propria and submucosa (figure 11), with extension to the mucosa and/or serosa. The tumour grows in solid sheets along with pseudopapillary, alveolar and nested patterns. The neoplastic cells exhibit a predominantly epithelioid appearance with round or oval and occasionally spindled nuclei and variable amounts of clear to lightly eosinophilic cytoplasm. Recently, there has been a case report of an oncocytic variant of malignant GNET as a previously undescribed potential diagnostic pitfall.99 The nuclei of GNET are usually centrally located and vesicular with variably prominent nucleoli, intranuclear cytoplasmic inclusions and easily identifiable mitotic figures. Extensive areas of tumour necrosis are seen. The most distinctive feature is the presence of scattered and unevenly distributed osteoclast-like multinucleated giant cells with the neoplastic cells. Soft tissue CCS type of neoplastic giant cells are absent.91–93

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Low-power photomicrograph depicting a gastrointestinal neuroectodermal tumour centred within the muscularis propria and submucosa (A). The tumour grows in solid sheets (B), and the neoplastic cells exhibit a predominantly epithelioid appearance with round or oval nuclei and clear to lightly eosinophilic cytoplasm (C). Occasional osteoclast-like multinucleated giant cells are scattered among the tumour cells (D).
Immunohistochemistry and ancillary studies
The immunohistochemical profile of GNETs suggests a primitive neural phenotype, characterised by S100 protein and SOX10 positivity, while being negative for melanocyte-specific markers, including HMB45, Melan A, tyrosinase and MiTF. Additionally, the neoplastic cells are diffusely positive for vimentin and are variably positive for CD57, CD56, synaptophysin, NSE and NB84. The neoplastic cells are also negative for DOG1, c-kit, keratins, smooth muscle actin and desmin.91–93 95 Ultrastructurally, GNETs show features of neural differentiation, including clear vesicles resembling synaptic bulbs, multiple interdigitating cell processes containing dense-core granules and lacks melanosomes.
Genetically, GNET is characterised by rearrangements of EWSR1 or FUS genes with CREB1 or ATF1, similar to CCS.91–95
Treatment and prognosis
GNET represents a highly malignant neoplasm with a poor prognosis and aggressive clinical behaviour including local recurrence, lymph node or visceral metastases, or death from disease, early or at the time of diagnosis in almost all reported cases. The possible role of adjuvant therapies in the treatment of GNET is unclear.93 95 97
Differential diagnosis
Awareness of the existence and diagnostic criteria of GNET by pathologists is crucial to avoid misdiagnosis of other histological mimics that come in its differential. These include CCS, MM, GIST, MPNST and monophasic synovial sarcoma. The histological features of GNET and CCS overlap including a nested growth pattern, the presence of clear to lightly eosinophilic cytoplasm, S100 protein expression and similar molecular translocations. But GNETs show certain differentiating features including pseudopapillary and sheet-like growth patterns, osteoclast-like giant cells, instead of Touton-type neoplastic giant cells seen in CCS, in addition to absence of specific melanocytic markers in GNET.
GNET may be distinguished from MM by virtue of the presence of osteoclast-like giant cells and the absence of immunohistochemical expression of specific melanocytic markers such as HMB45 and Melan A. GIST differs histologically from GNET and is positive for DOG1 and CD117. Monophasic synovial sarcoma expresses epithelial markers and TLE1, is negative for SOX10, and demonstrates a t(X;18) translocation, unlike GNET. MPNST usually does not show diffuse strong reactivity for S100 protein and lacks the genetic translocation involving EWSR1 gene. The oncocytic variant of GNET can be confused with malignant GCT as both tumours exhibit S100 protein and are SOX10 positive; however, awareness of this entity along with FISH testing to detect the characteristic translocation can help distinguish these entities.
Gastrointestinal autonomic nerve tumour
The first report of gastrointestinal autonomic nerve tumour (GANT) (in a 61-year-old man with a distal jejunal tumour) was published in 1984 by Herrera and colleagues.100 Based on the ultrastructural features, the authors considered the tumour to be a malignant enteric plexus tumour and termed it ‘plexosarcoma’. The term GANT was adopted later as suggested by Walker and Dvorak in 1986.101 Histogenetically related to the GI autonomic nervous plexus, GANT is not now regarded as a separate entity, but rather a variant of GIST (the only distinction is the electron microscopic findings).102 103 There are no clinical or treatment differences or prognostic benefit in subclassifying a lesion as GANT. However, in the interest of completeness, GANT is mentioned here.
Malignant peripheral nerve sheath tumour
Clinical features
MPNSTs are malignant tumours of ectomesenchymal origin, arising from a peripheral nerve, or in extraneural tissue showing nerve sheath differentiation, and are generally tumours of adulthood. They are the sixth most common type of soft tissue sarcoma, with an incidence of 0.1/100 000 per year.104 105
Approximately 50% of all MPNST cases arise sporadically, whereas the remainder are observed in patients with NF1, who carry an estimated 8%–13% lifetime risk of developing MPNST.106 In addition, an estimated 3%–10% of all patients with MPNST have a clinical history of prior radiation exposure, with development of MPNST after a latent period of >15 years.107
Most MPNSTs are located along major nerve trunks, commonly arising on the body trunk, extremities, head, neck and paravertebral regions. GI MPNSTs are rare entities, with only a handful of cases reported in the literature, including in the oesophagus,108 stomach,109 110 small intestine,111–115 large intestine116–118 and rectum.119 In the GIT, the disease is thought to arise from the Auerbach plexus, generally remaining in the submucosa, but exophytic or intraluminal growth can occur.114 Other uncommonly reported sites of MPNST include pancreas,120 bile duct121 and liver.122–124 The majority of small intestinal MPNSTs reported presented with intestinal obstruction due to intussusception.
Microscopic findings
Histologically, most MPNSTs are relatively monomorphic spindle cell malignancies, frequently composed of fascicles of alternating cellularity, whorls, palisades or rosette-like arrangements, with subendothelial accentuation of tumour cells and large areas of geographic necrosis. In general, the great majority of MPNSTs are high grade, spindle cell neoplasms showing unequivocal cytological atypia, high mitotic activity and necrosis. Heterologous differentiation in the form of bone and/or cartilage, or less commonly smooth muscle, skeletal muscle and well-formed glands occurs occasionally, particularly in patients with NF1.125–127 Heterologous elements may portend an even poorer prognosis; MPNSTs demonstrating skeletal muscle differentiation (malignant Triton tumours) are particularly aggressive and associated with a poor prognosis.128
The main recognisable benign precursor to MPNST is neurofibroma, in particular the plexiform type in the setting of NF1. Malignant transformation of schwannoma into MPNST is a much rarer phenomenon.129 MPNSTs may also arise in GNs and ganglioneuroblastomas.130
Recently, a consensus group designated a new category of ‘atypical neurofibromatous neoplasm of uncertain biological potential’ (ANNUBP) for tumours that show some worrisome features of malignant transformation, but histologically fall short of MPNST. ANNUBP has been defined as a Schwann cell neoplasm with at least two of four of the following features: loss of neurofibroma architecture, hypercellularity, cytological atypia and mitotic index >1/50 HPFs and <3/10 HPFs. They recommend that a diagnosis of MPNST, low grade, should be made when a tumour has features of ANNUBP, but with a mitotic index of 3–9/10 HPFs and no necrosis. Finally, the consensus group stated that a tumour should be designated as a MPNST, high grade, when there are histological features of MPNST, with at least 10 mitotic figures/10 HPFs or 3–9 mitotic figures/10 HPFs, combined with necrosis.131
Immunohistochemistry and ancillary studies
Weak, patchy S100 protein immunoreactivity is detected in 50%–60% of MPNSTs but it is also widely expressed in a range of other tissues and tumour types.132 Strong diffuse S100 staining nearly always excludes a diagnosis of MPNST.128 Other antigens useful in determining nerve sheath differentiation include Leu-7 and myelin basic protein.133 A recent study showed that neurofibromin expression was lost in both NF1-associated and sporadic MPNSTs, while it was retained in other spindle cell tumours.134 MPNSTs may also show expression of SOX10, low molecular weight cytokeratins, EMA and CD34.
No specific chromosomal rearrangements have been uncovered in MPNST, but a complex karyotype is usually present.135 In addition, molecular alterations including EGFR amplification and deletions of NF1 or CDKN2A (p16) are supportive of a diagnosis of MPNST in the appropriate setting.136 137 Mutations in TP53 have been shown to be associated with MPNST development, with a reported incidence of TP53 aberrations in MPNST ranging from 25% to 80%.138 139 A recently discovered marker of MPNST is the loss of expression of trimethylated histone 3 at lysine residue 27 (H3K27me3), resulting from loss-of-function mutations in EED and SUZ12 genes.140
Prognosis
Criteria for the histological grading of MPNST are controversial and there is a paucity of data in the literature regarding prognostic factors and long-term outcome in MPNST, but reported 5-year survival rates range from 35% to 50%141 and disease-specific survival rates at 10 years range from 7.5% to 31.6%.142 It is thought that MPNST of the bowel has a far worse prognosis than other soft tissue sarcomas.111
Factors that are reported to predict recurrence include anatomic site, tumour size (≥10 cm) and adequacy of resection margins. Factors that are reported to predict metastases include a lack of S100 staining, tumour size (≥10 cm) or tumours that are American Joint Committee on Cancer stage III.141 143 Studies of MPNSTs at all sites have suggested that elevated Ki67 expression (>25%) is associated with decreased survival.141 144 Several studies have revealed that TP53 and MDM2 may promote metastasis development and thereby contribute to malignant progression in MPNST,145 with recently published data in the USA showing that nuclear TP53 expression is significantly associated with a worse MPNST-specific survival outcome141 (table 2).
Prognostic factors for malignant peripheral nerve sheath tumour
Differential diagnosis
The differential diagnosis of MPNST is with a variety of other sarcomas, including synovial sarcoma and leiomyosarcoma. Low-grade variants of MPNSTs may resemble cellular neurofibromas (criteria for distinction outlined previously). One of the most useful distinctions from benign Schwann cell tumours is the diminished or even absent S100 staining in MPNST.133 146 MPNSTs, and indeed schwannomas, that were reported in the literature prior to the advent of CD117 as a diagnostic marker, are invariably GISTs. Thus, several of the earlier publications for both of these entities are of limited strength.
Neurofibromatosis type 1 and the GIT
NF1 is an autosomal-dominant condition that, affecting 1 in 3000 live births, represents the most common human cancer genetic predisposition syndrome. It is almost completely penetrant, but demonstrates variable expressivity, a phenotypic heterogeneity that renders accurate prediction and early detection of clinical complications like malignant transformation challenging.
This syndrome is characterised by multiple areas of cutaneous hyperpigmentation (café-au-lait spots) and numerous neurofibromas. Other clinical features include axillary freckling, optic gliomas, iris hamartomas (Lisch nodules) and bone dysplasia. NF1 can also be associated with cardiovascular abnormalities, learning deficiencies and the development of a variety of malignancies (including ependymomas, lymphomas, myeloid leukaemia, Wilms tumour, phaeochromocytomas, rhabdomyosarcoma, MPNST and carcinoids).2
The frequency of intra-abdominal (GI or retroperitoneal) manifestations of NF1 reported in studies varies greatly, ranging from 5% to 25%.1 147 GI manifestations usually arise during mid-life or later, generally after the appearance of the cutaneous manifestations of the disease. Peripheral nerve sheath tumours of different histological subtypes may develop in the GIT of patients with NF1, occurring at any site from the oesophagus to the anorectum.1 147 Four main lesions are encountered: neurofibroma, DG, GIST and MPNST. Several recent studies have described a high rate of GISTs in patients with NF1; these data suggest that GISTs are in fact a more common GI manifestation of NF1 than neurofibromas.148 149 Recently, Agaimy et al described a series of NF-1-related inflammatory GI polyps,150 which have a wide range of histological appearances, ranging from a predominantly inflammatory pattern to a fibrovascular granulation tissue-like and juvenile-like appearance.
The genotypic hallmark of NF1 involves mutations to, or other loss of, the gene NF1 (a tumour suppressor gene) on the long arm of chromosome 17, which encodes the tumour suppressor protein neurofibromin.
Conclusion
The realm of peripheral nerve sheath tumours in the gastroenteropancreaticobiliary tract is currently a dynamic, evolving area of surgical pathology. Morphological variability of these tumours is wide, although overlap between some entities also occurs and thus they can engender some difficult differential diagnoses.
In summary, this review is an attempt to simplify the diagnosis of this varied group of benign as well as malignant, common and uncommon neural neoplasms in the GI tract by systematically discussing these entities along with the helpful clinical and histopathological features and differential considerations.
Take home messages
Neural lesions of the gastrointestinal and pancreaticobiliary tract can present significant diagnostic challenges both for the pathologist and the clinician, in part because of overlapping clinical presentations as well as histopathological features.
Awareness of the wide spectrum of neural lesions that can manifest in the gastrointestinal and pancreaticobiliary tracts is crucial for accurate diagnosis and optimal patient management.
We review the spectrum of both benign and malignant neural tumours in the gastroenteropancreaticobiliary tract, with the goal of providing a practical simplistic approach for practising surgical pathologists.
References
Footnotes
Handling editor Tahir S Pillay.
Contributors All authors contributed equally.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.