Article Text

Abstract
Fumarate hydratase (FH), encoded by the FH gene, is an enzyme which catalyses the conversion of fumarate to L-malate as part of the tricarboxylic acid cycle. Biallelic germline mutations in FH result in fumaric aciduria, a metabolic disorder resulting in severe neurological and developmental abnormalities. Heterozygous germline mutations in FH result in hereditary leiomyomatosis and renal cell carcinoma, a cancer predisposition syndrome. FH deficiency has multiple oncogenic mechanisms including through promotion of aerobic glycolysis, induction of pseudohypoxia, post-translational protein modification and impairment of DNA damage repair by homologous recombination. FH-deficient neoplasms can present with characteristic morphological features that raise suspicion for FH alterations and also frequently demonstrate loss of FH immunoreactivity and intracellular accumulation of 2-succinocysteine, also detected by immunohistochemistry.
- neoplastic syndromes
- hereditary
- genetics
- oncogenes
Statistics from Altmetric.com
Introduction
The FH gene encodes for the protein fumarate hydratase (FH), a key enzyme in the tricarboxylic acid (TCA) cycle.1 Pathogenic FH mutations and the resultant deficiency in FH function result in a myriad of metabolic and cell signalling alterations, many of which promote oncogenesis, which explains the association between germline mutations in FH and the increased risk for certain neoplasms.
Gene and protein structure
FH is located on chromosome 1 (1q42.3-q43) and is composed of 10 exons.1 2 The FH protein is a 510-amino acid structure with three key domains: a N-terminal lyase 1 domain, a C-terminal fumarase C domain and a central domain which interacts with other FH monomers.1 3 In its functional form, FH exists as a homotetramer4 and can be localised in the mitochondria or the cytosol. Both mitochondrial and cytosolic FH are encoded by nuclear DNA, with the differential localisation mediated by alternative transcription products, one of which produces a precursor protein with an N-terminal mitochondrial localisation signal.1 5
Physiological role
Within the mitochondria, homotetrameric FH catalyses the reversible conversion of fumarate to L-malate as part of the TCA cycle, a critical pathway in cellular metabolism which processes substrates for the production of ATP through oxidative phosphorylation.1 4
Cytosolic FH can be translocated to the nucleus to participate in DNA damage repair.6 Upon localisation to the nucleus, FH catalyses fumarate production, with the resultant increased fumarate concentration inhibiting the histone demethylase KDM2B and promoting repair of double-strand breaks by non-homologous end-joining.6 7 In addition, cytosolic FH is crucial for proper functioning of the urea cycle.8 In cases of FH deficiency, excess cytosolic fumarate leads to accumulation of argininosuccinate, and FH-deficient cells become acutely dependent on exogenous arginine due to disruption of normal urea cycle metabolism.1 8
Role in disease
Fumaric aciduria
Biallelic germline pathogenic mutations in FH are very rare and result in fumaric aciduria, a condition that occurs due to a systemic inability to effectively convert fumarate to malate.9 Affected patients have high levels of urinary fumarate and detectable aberrations in serum levels of other TCA cycle metabolites.3 10 The associated phenotype is severe with multiple dysmorphic features, hypotonia and profound, global developmental deficits presenting in infancy; only rare survival past childhood has been reported.3 10
FH deficiency and tumour development
Putatively oncogenic missense mutations are most common in exons 4, 5, 7 and 8.11 FH-deficient cells lose the capacity to metabolise fumarate and intracellular levels consequently increase, resulting in a series of compensatory metabolic shifts.1 For example, mitochondrial oxidative phosphorylation is reduced while aerobic glycolysis, glutamine catabolism and major anabolic pathways are upregulated; these are features of the Warburg effect, one of the metabolic hallmarks of malignancy.1 12 13
In addition to its impact on TCA cycle dysregulation, increased intracellular fumarate is capable of inducing other oncogenic cellular alterations, justifying its classification as an oncometabolite.14 One of the major changes associated with increased fumarate is constitutive stabilisation of hypoxia inducible factor (HIF)-1α and HIF-2α.15 Under normal physiological conditions, HIF-1α and HIF-2α are constitutively hydroxylated by a prolyl hydroxylase, which results in their downstream ubiquitylation and subsequent proteasomal degradation.16 Under hypoxic conditions, these proteins are stabilised and capable of inducing transcription of key genes involved in angiogenesis and cell growth.17 Elevated intracellular fumarate is believed to inhibit α-ketoglutarate-dependent dioxygenases including prolyl hydroxylase and thereby prevent ubiquitylation and degradation of these HIF-family proteins.15 16 18 19 Moreover, FH deficiency is associated with increased reactive oxygen species which can also inhibit prolyl hydroxylase activity.18 Additional evidence for the importance of this signalling pathway has been found in FH-deficient uterine leiomyomata (discussed below), which have higher microvessel density and increased expression of vascular endothelial growth factor than their non-FH-deficient counterparts.20 However, the relative contribution of these mechanisms to HIF-family activity and its contribution to oncogenesis in FH-deficient cells remains a matter of controversy.15
Increased fumarate levels also result in post-translational succination of cysteine residues on a number of different proteins. One of these proteins is KEAP1 which normally induces degradation of NRF2.15 NRF2 upregulation leads to increased intracellular ferritin and the resultant activation of the transcription factor FOXM1 which promotes cellular proliferation as well as an antioxidant response phenotype.1 15 21 Other proteins preferentially succinated in FH-deficient cells include those involved in metabolism and the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodelling complex.14 Positive immunohistochemical staining for 2-succinocysteine (2-SC) is a marker of protein succination, and has been shown to be a reliable marker for tumourous FH deficiency (figure 1).22

2-succinocysteine (2-SC) immunohistochemistry. Increased intracellular fumarate levels as a result of fumarate hydratase deficiency leads to increased protein succination which can be detected through immunohistochemistry for 2-SC. In this example of a fumarate hydratase-deficient uterine leiomyoma, the tumour cells show abnormal diffuse cytoplasmic granular expression of 2-SC, in contrast to background endothelial, stromal and inflammatory cells which show no expression. Image magnification ×400.
In addition, dysregulation of normal FH activity has been shown to impair DNA repair by homologous recombination through alterations in histone methylation.23 An additional more recently described oncogenic effect of FH deficiency is the promotion of epithelial-to-mesenchymal transition (EMT) via downregulation of the miR-200 family of micro-RNAs and subsequent upregulation of an EMT-associated signature.24
The main consequences of FH deficiency and increased fumarate levels are shown in figure 2.

Consequences of increased fumarate levels. Fumarate hydratase catalyses the conversion of fumarate to malate in the tricarboxylic acid cycle (A). Inhibition of this enzyme causes increased levels of fumarate which inhibit oxidative phosphorylation in the mitochondria. Separately, increased levels of cytosolic and nuclear fumarate result in upregulation of HIF-family transcription factors, protein succination and aberrant DNA repair, among other impacts (B). Created with BioRender.com. 2-SC, 2-succinocysteine; HIF, hypoxia inducible factor.
Hereditary leiomyomatosis and renal cell carcinoma
Heterozygous, pathogenic germline mutations in FH are associated with an autosomal dominant cancer predisposition syndrome called hereditary leiomyomatosis and renal cell carcinoma (HLRCC).25 This condition is associated with frequent and multiple cutaneous leiomyomata and aggressive RCCs and, in females, with early-onset, symptomatic uterine leiomyomata.25 In keeping with the role of FH as a tumour suppressor, all somatic tissues in HLRCC patients have only one functional copy of the FH gene; mutation or loss of this remaining copy results in the ‘second hit’ that initiates tumourigenesis.15 Pathogenic FH mutation can be reliably detected in up to 100% of patients meeting clinical criteria for the syndrome; large exonic deletions, which may require multiplex ligation-dependent probe amplification to detect, appear to occur only rarely in patients with HLRCC.25 26 There is evidence to suggest that truncating mutations may be associated with a milder phenotype than missense mutations with the latter postulated to have a dominant-negative impact that actively inhibits homotetramer formation in at least some cases.27
FH-deficient neoplasms
FH-deficient RCC
RCCs with FH deficiency occur almost exclusively in the setting of HLRCC however, FH-deficient RCC can also arise sporadically secondary to loss of function in both FH alleles.28–30 FH-deficient RCC arises in approximately 15%–30% of patients with HLRCC and in the syndromic setting, these tumours are associated with an early age of onset and an aggressive clinical course with one study finding that 50% of patients died of disease after a median follow-up of only 16 months.25 31 32
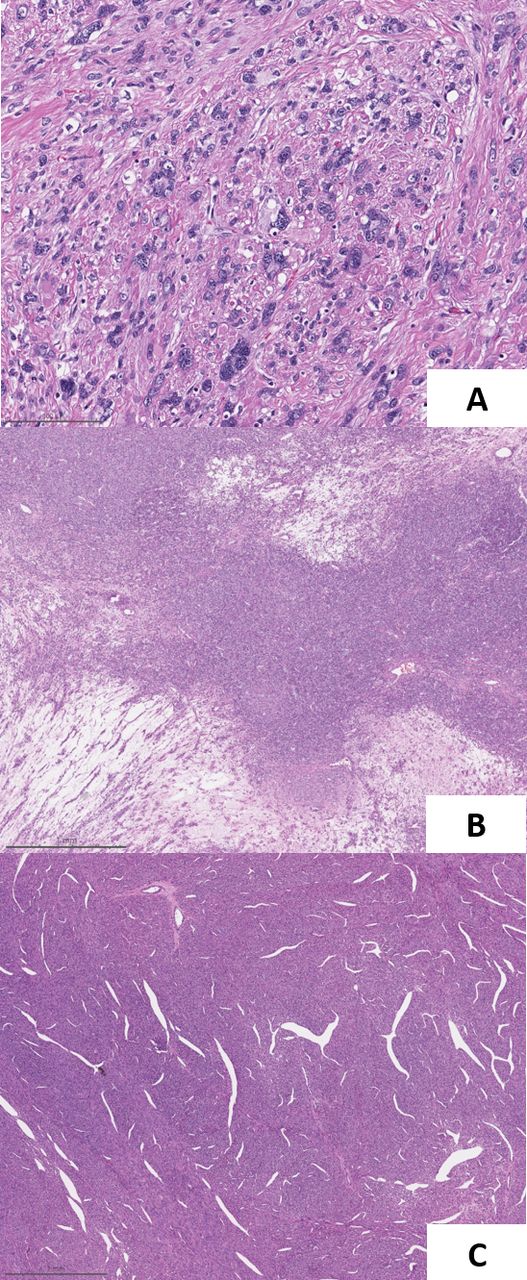
Histologically, the tumours frequently display multiple architectural patterns within the same lesion, with papillary architecture the most common but by no means ubiquitous.31 Also characteristic are large, eosinophilic, ‘inclusion-like’ nucleoli, though their presence is often variable31 (figure 3). Less common morphological variants include the characteristic low-grade, oncocytic cytology more typically associated with succinate dehydrogenase (SDH)-deficient RCC, and tubulocystic architecture with poorly differentiated foci.33 34 Immunoreactivity for FH is frequently lost but can be retained in a subset of cases in which a non-functional protein is expressed, while immunohistochemistry for 2-SC can also be a useful ancillary study.28 31 32 35

Fumarate hydratase (FH)-deficient renal cell carcinoma. This example shows nested and solid architecture, abundant eosinophilic cytoplasm and conspicuous single macronucleoli (A). Loss of FH immunoexpression can be a useful ancillary technique for confirming this diagnosis; note how background endothelial cells act as an internal control (B). Image magnification ×200x.
Historically, these FH-deficient RCCs were classified as type 2 papillary RCCs, given their nuclear features and frequent papillary architecture.25 In The Cancer Genome Atlas Research Network study of papillary RCC, FH-deficient cases clustered with the CpG island methylator phenotype subgroup of tumours, characterised by multiple metabolic alterations and an aggressive clinical course.36 However, HLRCC-associated RCC is now a distinct subtype of RCC in its own right, a reflection of the unique aetiology and clinicopathological characteristics associated with this entity.28 29
Uterine leiomyomata
FH deficiency is a rare finding in unselected populations of patients with uterine leiomyomata and unlike FH-deficient RCCs, a higher proportion of these FH-deficient uterine tumours are not associated with underlying germline mutation.37–39 In the setting of HLRCC, affected women characteristically develop early-onset and symptomatic uterine leiomyomata which often require early surgical intervention.35 37
Similar to FH-deficient RCCs, FH-deficient uterine leiomyomata are associated with certain morphological features that may raise suspicion as to their underlying pathogenesis.40 FH-deficient leiomyomata are more likely than other leiomyomata to present with symplastic-type nuclear atypia, misinterpretation of which may result in erroneous classification as leiomyosarcoma.37 38 41 More specific features include hemangiopericytoma-like (‘staghorn’) blood vessels, ‘alveolar-type’ stromal oedema, eosinophilic cytoplasmic globules and large, eosinophilic nucleoli (figure 4); none of these features in isolation show perfect diagnostic accuracy, but their conspicuous presence within a tumour should prompt further consideration that one is dealing with an FH-deficient tumour, particularly in a young patient.35 As with FH-deficient RCC, immunohistochemistry for FH and 2-SC can be helpful ancillary tools for diagnosis although of note, morphology and immunohistochemistry alone cannot distinguish between a uterine tumour with somatic versus germline FH mutation. As such, consideration should be given to germline testing when characteristic morphology is identified, even in the absence of supportive immunohistochemical results, as it has been shown that neither FH or 2-SC immunohistochemistry correlates perfectly with molecular testing for FH mutation.35 42

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fumarate hydratase (FH)-deficient uterine leiomyoma. Characteristic features of FH-deficient uterine leiomyomata include marked nuclear atypia, single prominent nucleoli with perinucleolar clearing, hyaline globules (A), ‘alveolar-type’ stromal oedema (B) and hemangiopericytomatous (‘staghorn’) blood vessels (C). These morphological features may be seen in tumours with somatic FH alterations or in the context of germline FH changes. Image magnification ×200 (A) and ×20 (B, C).
FH-deficient leiomyomata of the uterus are not specifically associated with an increased risk of malignancy, however, their identification is extremely important as by recognising the characteristic phenotype and/or confirming FH-deficiency by IHC or molecular testing, pathologists can recommend germline testing for FH mutations so as to identify patients that may benefit from routine screening for HLRCC-associated RCC.40
Cutaneous leiomyomata
Cutaneous leiomyomata, believed to derive from the arrector pili muscles in hair-bearing skin, are not always associated with FH deficiency and occur outside of the syndromic setting however, when multiple and widespread, are often the most conspicuous manifestation of HLRCC.27 In the syndromic setting, affected patients often have multiple grossly apparent papules or nodules27 43 and of note, they occur with higher penetrance in men.27 Morphologically, cutaneous leiomyomata resemble leiomyomata of other locations and, unlike HLRCC-associated RCCs and uterine leiomyomata, do not have any known distinctive histological features.44 45 While benign, these lesions can cause significant morbidity and can be painful, particularly with changes in temperature.43 Like FH-deficient uterine leiomyomata, the presence of multiple cutaneous leiomyomata should alert clinicians to test for germline FH mutation, so that appropriate screening for RCC can be initiated.44 Immunohistochemistry for 2-SC can be a useful adjunct for detection of FH-deficiency in these tumours, with the utility of immunohistochemistry for FH considered to be more controversial.44 45
Other
FH-deficiency is associated with some of the same downstream effects on HIF-1α signalling as SDH family mutations and while SDH deficiency is associated with a different spectrum of tumour predispositions compared with HLRCC, a small amount of overlap does exist.1 15 For example, FH deficiency as a result of germline mutations has been implicated as a rare cause of malignant paragangliomas and pheochromocytomas.46 Other FH-deficient tumours include Leydig cell tumours of the testis and rare bladder and breast carcinomas that have been identified in patients with HLRCC.27 47 Uterine leiomyosarcoma has been controversially associated with HLRCC, with a study out of Finland reporting a substantially increased risk in these patients, but other large cohort studies have failed to identify any FH-deficient leiomyosarcomas.37 38 48
Take home messages
Fumarate hydratase (FH), encoded by the FH gene of chromosome 1, catalyses the conversion of fumarate to L-malate as part of the tricarboxylic acid cycle, and also plays roles in the urea cycle and DNA repair.
Biallelic germline mutations in FH result in a severe inborn disorder of metabolism, fumaric aciduria, that is characterised by profound neurological and developmental deficits.
Heterozygous germline loss-of-function mutations in FH cause hereditary leiomyomatosis and renal cell carcinoma, which predisposes to an aggressive variant of renal cell carcinoma and other tumours.
Some FH-deficient neoplasms have distinct morphological features but immunohistochemistry and/or molecular testing are often required to secure a definitive diagnosis.
Ethics statements
Patient consent for publication
References
Footnotes
Handling editor Runjan Chetty.
Contributors REZ and AH worked cooperatively and contributed equally to this manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.